
Phosphorylation controls the oligomeric state of She2 and mRNA localization in yeast
- Department of Biochemistry and Molecular Medicine, Université de Montréal, Montréal, Quebec H3C 3J7, Canada
- Corresponding author: p.chartrand{at}umontreal.ca
-
↵1 These authors contributed equally to this work.
Abstract
Messenger RNA (mRNA) localization is an important mechanism controlling local protein synthesis. In budding yeast, asymmetric localization of transcripts such as ASH1 mRNA to the bud tip depends on the She2 RNA-binding protein. She2 assembles as a tetramer to bind RNA, but the regulation of this process as part of the mRNA locasome is still unclear. Here, we performed a phosphoproteomic analysis of She2 in vivo and identified new phosphosites, several of which are located at the dimerization or tetramerization interfaces of She2. Remarkably, phosphomimetic mutations at these residues disrupt the capacity of She2 to promote Ash1 asymmetric accumulation. A detailed analysis of one of these residues, T109, shows that a T109D mutation inhibits She2 oligomerization and its interaction with She3 and the importin-α Srp1. She2 proteins harboring the T109D mutation also display reduced expression. More importantly, this phosphomimetic mutation strongly impairs the capacity of She2 to bind RNA and disrupts ASH1 mRNA localization. These results demonstrate that the control of She2 oligomerization by phosphorylation constitutes an important regulatory step in the mRNA localization pathway.
Keywords
INTRODUCTION
Messenger RNA localization contributes to the post-transcriptional regulation of gene expression by controlling the synthesis, in space and time, of specific proteins (Das et al. 2021). This process has been observed in multiple organisms, tissues and cell types, and can involve a significant proportion of the transcriptome in some organisms (Lécuyer et al. 2007; Das et al. 2021). The budding yeast Saccharomyces cerevisiae is an established model organism for studying this question since dozens of transcripts are transported from the mother cell and localized to the budding daughter cell (Shepard et al. 2003; Oeffinger et al. 2007; Pizzinga et al. 2019; Chaudhuri et al. 2020). One of these transcripts, ASH1 mRNA, is transported to the bud during late anaphase and has been extensively studied (Long et al. 1997; Takizawa et al. 1997). The journey of ASH1 mRNA to the bud tip starts with the cotranscriptional interaction of She2, the main RNA-binding protein (RBP) involved in bud-localization of mRNAs, with four cis-acting elements (or zipcodes) along the mRNA sequence (Chartrand et al. 1999; Shen et al. 2010). She2 helps recruit the translational repressor Puf6 on ASH1 cotranscriptionally, via a common partner, Loc1 (Shahbabian et al. 2014). Khd1, another translational repressor, is also loaded on this transcript in the nucleus (Irie et al. 2002). After transcription, the ASH1 mRNP complex is exported to the cytoplasm, where She3 replaces Loc1 to assemble a stable mRNP competent for localization (Müller et al. 2009; Niedner et al. 2013). The transport of the mRNP requires the type V myosin Myo4 and the actin cytoskeleton until the complex reaches the bud tip, where its local translation occurs (Munchow et al. 1999; Takizawa and Vale 2000).
She2 is the key RBP responsible for the localization of transcripts at the bud tip (Bohl et al. 2000; Long et al. 2000). The She2 polypeptide is a noncanonical RBP, which assembles into a tetramer and contains two basic-rich RNA-binding domains that can bind two independent RNA localization elements (Edelmann et al. 2017). The oligomerization of She2 is essential for its interaction with Loc1 and She3, which help stabilize the RNA–protein complex in the nucleus and cytoplasm, respectively (Müller et al. 2011; Niedner et al. 2013). However, She2 oligomerization decreases its capacity to bind the importin-α Srp1, which promotes the nuclear import of She2 via its interaction with a noncanonical nuclear localization sequence at the carboxyl terminus (Shen et al. 2009). How these various interactions are regulated remains unclear.
While She2 has been reported to be phosphorylated in vivo (Gonsalvez et al. 2003), only one phosphorylated residue (S166) has been identified so far in a large-scale phosphoproteomic study (Smolka et al. 2007). Herein, we used phosphoproteomic analysis to identify the phosphorylated residues in She2 in vivo. We identified several novel phosphosites that impact the capacity of She2 to promote the asymmetric accumulation of Ash1. Interestingly, some of these phosphosites are present at the dimerization and tetramerization interfaces of She2. Focusing on T109, we show that the phosphomimetic mutant T109D inhibits She2–She2 interaction, and decreases the interaction of She2 with its partners Srp1, She3, and ASH1 mRNA. Interestingly, the T109D mutation significantly reduces the expression of the She2 protein. Together, our results show that the control of She2 oligomerization by phosphorylation represents a novel mechanism regulating mRNA localization in budding yeast.
RESULTS AND DISCUSSION
Identification of phosphorylated amino acid residues in She2
To determine if She2 is a phosphoprotein, we used the Pro-Q Diamond phosphoenrichment resin that binds specifically to phosphoproteins (Kristjansdottir et al. 2008). The passage of a yeast extract on a Pro-Q column, followed by washes and elution, revealed She2-myc in the eluate (Fig. 1A), suggesting that a fraction of She2 is phosphorylated in vivo. A positive control, phospho-S6 protein, was also detected in the eluate (Fig. 1A). To validate the specificity of the Pro-Q resin for binding phosphorylated She2, yeast extracts were treated with phosphatase before loading on the column. Compared with She2-myc from untreated yeast extract, which binds to the phosphoenrichment resin, She2-myc from phosphatase-treated yeast extract was not retained on the Pro-Q column (Fig. 1B). Notably, a faster migrating band of unknown origin appears when extracts are treated with phosphatase. Still, it remains possible that She2 interaction with a phosphorylated protein may explain its binding to the Pro-Q resin (Kristjansdottir et al. 2008). Indeed, She2 interacts with She3, which is a known phosphoprotein (Landers et al. 2009). To eliminate this possibility, yeast extracts were treated with urea to denature proteins before loading on the Pro-Q column. As shown in Figure 1C, urea treatment did not impact the binding of She2-myc to the column, suggesting that She2 directly interacts with the Pro-Q resin. Altogether, these results show that a fraction of She2 is phosphorylated in budding yeast.
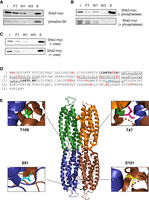
Identification of phosphorylated residues in She2. (A) Binding of She2-myc on the Pro-Q phosphoprotein enrichment column. Detection of She2-myc by western blot from the whole lysate (I: input), flow-through (FT), the first wash (W1), third wash (W3), and eluate (E) from the Pro-Q column. Phosphorylated ribosomal protein S6 (phospho-S6) was used as a positive control (bottom). (B) Binding of She2 to the Pro-Q column depends on its phosphorylation state. Yeast extracts were treated with Fast phosphatase (+phosphatase) or incubated without phosphatase (−phosphatase) prior to binding to the Pro-Q column. Detection of She2-myc was performed as in A. (C) Denaturation of She2 with urea does not reduce its binding to the Pro-Q column. Yeast extracts were treated with 8M urea (+urea) or not (−urea) prior to binding to the Pro-Q column. Detection of She2-myc was performed as in A. (D) Sequence of She2 with phosphorylated amino acids identified by LC–MS/MS colored in red. Amino acids at the dimerization interface of She2 are underlined. Amino acids in the dimer-dimer interface of She2 are in bold. Amino acids in blue are phosphosites identified in previous studies. (E) Structure of the She2 tetramer. The locations of the phosphorylated residues T47, S91, S101, and T109 in the 3D structure are highlighted. She2 3D structure was generated with the PyMOL software using the 5M0J structure from the PDB database.
To identify phosphorylated residues in She2, a yeast strain expressing a GST-She2 fusion protein under the galactose-inducible GAL1 promoter was generated. GST-She2 is known to fold into a functional protein, as it binds both ASH1 RNA zipcodes and She3 (Bohl et al. 2000; Gonsalvez et al. 2003; Olivier et al. 2005). This strain allowed sufficient induction of GST-She2 for subsequent purification of the full-length protein using glutathione beads, SDS/PAGE and LC/MS–MS analysis. Three independent phosphopeptide analyses revealed several phosphosites in She2 (Fig. 1D; Supplemental Tables 1–3). Interestingly, some phosphosites are present at the dimerization interface between She2 monomers, such as Y65, S91, S101, and T109 (Fig. 1D,E). Another phosphosite, T47, is located at the interface between two She2 dimers (Fig. 1E; Müller et al. 2009). Finally, two phosphorylated residues (S217 and S224) are in the nuclear localization sequence (NLS) of She2 (Shen et al. 2009). These results suggest that the She2 monomer may be a substrate for phosphorylation by kinases at residues Y65, S91, S101, and T109, which are nonaccessible in the She2 dimer/tetramer.
Phosphorylation of She2 impacts Ash1 asymmetric distribution
To identify phosphorylated residues that regulate the activity of She2, we generated phospho-null (S/T to A; Y to F) and phosphomimetic (S/T to D) mutations at specific phosphoresidues. The myc-tagged She2 protein was expressed from its endogenous promoter on a centromeric plasmid. The activity of the mutated She2 proteins was tested in a yeast genetic assay to assess the asymmetric distribution of the Ash1 protein and its capacity to repress the HO promoter. In the K5547 strain, in which the ADE2 gene is under the control of the HO promoter and that contains a deletion of the SHE2 gene, a symmetric distribution of Ash1 between mother and daughter cells (due to defective ASH1 mRNA localization) leads to repression of the ADE2 gene and poor growth on plates lacking adenine (−Ade). Expression of WT She2 or a functional She2 mutant in this strain restores ASH1 mRNA localization and asymmetric accumulation of Ash1 in the daughter cell and rescues the expression of the ADE2 gene in the mother cell, allowing growth on −Ade plates (Jansen et al. 1996).
Using serial dilutions and spot assays, expression of wild-type She2 rescued the growth defect of the K5547 strain on a −TRP −ADE medium, while the expression of the She2 M5A mutant, which disrupts the NLS of She2 (Shen et al. 2009), or the empty vector YCPlac22, did not rescue cell growth on −TRP −ADE medium (Fig. 2A). The impact of mutations at phosphorylated residues S2, T47, Y65, S91, S101, T109, Y148, S217, and S224 on Ash1 asymmetric localization was tested in this genetic assay. As shown in Figure 2B, most mutations at phosphosites did not affect the capacity of She2 to promote Ash1 asymmetric distribution and growth on −TRP −ADE medium (see also Supplemental Figs. 1, 2). However, the expression of a T109A mutant only partially rescued the growth defect, while the expression of the phosphomimetic T109D mutant did not rescue growth (Fig. 2A,B). Interestingly, besides T109, phosphomimetic mutations at T47 (T47D) or S101 (S101D) did not rescue yeast growth on the selection medium, whereas the phospho-null mutant at T47 (T47A) or S101 (S101A) did, suggesting that phosphorylation of these residues also disrupts She2 activity (Fig. 2B; Supplemental Figure 1).
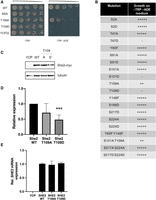
Specific phosphoresidues modulate the capacity of She2 to promote the asymmetric distribution of Ash1. (A) Genetic assay to assess Ash1 asymmetric distribution in She2 mutants. Serial dilutions of K5547 + YCP22-She2-myc, K5547 + YCP22-She2-M5A-myc, K5547 + YCP22-She2-T109A-myc, K5547 + YCP22-She2-T109D-myc, and K5547 + YCP22 empty. (B) Impact of various phospho-null and phosphomimetic mutants of She2 on the growth of K5547 on −ADE −TRP medium. (C) Western blotting of She2-myc WT, T109A, and T109D expression in the K5547 strain. YCP: K5547 + YCPlac22 empty. Tubulin was used as a loading control. (D) Quantification of She2 WT, She2 T109A, and She2 T109A expression in the K5547 strain. (***) P < 0.005. N = 3. (E) Relative expression of SHE2 mRNA quantified by RT-qPCR in K5547 strain expressing YCPlac22 (YCP), She2 WT, She2 T109A, or She2 T109D proteins. N = 2.
Expression of the wild-type and T109 mutated She2-myc proteins was assessed by western blot, which revealed a twofold decrease in the expression of the She2 T109D mutant compared to the wild-type or T109A She2 proteins (Fig. 2C,D). RT-qPCR analysis of SHE2 mRNA levels showed that the T109D mutation did not affect SHE2 mRNA expression (Fig. 2E), suggesting that a phosphomimetic mutation at T109 significantly reduces the accumulation of the She2 protein in budding yeast. Unlike T109D, expression of the S101D and T47D mutants of She2 was similar to that of the wild-type protein, even though these mutants also disrupt Ash1 protein asymmetric localization (Supplemental Fig. 1).
Since phosphomimetic mutations at S101 and T109 strongly impact the function of She2, and both residues are located at the dimerization interface between She2 monomers (Fig. 1E), we tested a phosphomimetic mutant at S166, which is also located at the dimerization interface (Fig. 1D) and was previously identified in a large-scale phosphoproteomic study (Smolka et al. 2007). Unlike S101D and T109D, expression of She2 S166D fully complemented the SHE2 knockout and these cells could grow on −TRP −ADE medium, like the cells expressing She2 WT (Fig. 2B).
Finally, double phospho-null or phosphomimetic mutants were also tested to explore the possible contribution of multiple phosphorylation events to the activity of She2. Double phospho-null mutations at Y65 Y148 or S217 S224 reproduced the same phenotype as single mutations at these residues (Fig. 2B). Similarly, a double phosphomimetic mutant at S217 S224 showed the same growth as She2 WT. However, the double mutation S101A T109A reproduced the same phenotype as the single T109A mutation (Fig. 2B), suggesting that these mutations do not have an additive impact on the function of She2.
Phosphomimetic mutation at Threonine 109 disrupts She2 dimerization and its interactions with Srp1 and She3
Our phosphoproteomics analysis revealed that phosphorylation at Threonine 109 (T109) is the most frequently found phosphorylated residue on She2. Indeed, it was the only phosphosite found in all three independent experiments, with 30 peptides containing this phosphorylated amino acid (Supplemental Tables 1–3). Since the phosphomimetic T109 She2 mutant disrupts Ash1 protein asymmetric distribution (Fig. 2A), the impact of a T109 phosphomimetic mutant on the function of She2 was explored. First, the effect of the mutations T109A or T109D on She2–She2 interaction was assessed in a yeast two-hybrid assay. As shown in Figure 3A, the T109A She2 mutation in both bait and prey led to a threefold reduction in β-galactosidase activity compared to the wild-type She2–She2 interaction. The impact of the T109D mutation on She2–She2 interaction is even more significant, with a nearly 20-fold decrease in interaction for the homodimer She2-T109D:She2-T109D or for the formation of the heterodimer She2 WT:She2-T109D (Fig. 3A).
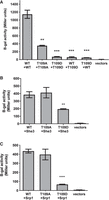
Phosphomimetic mutation at T109 inhibits the oligomerization of She2 and its interaction with cofactors Srp1 and She3. (A) Yeast two-hybrid assay to detect She2–She2 interaction. Homo- or hetero-oligomerization of wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) was quantified by measuring β-galactosidase activity (N = 3). (**) P < 0.01; (***) P < 0.005. (B) Yeast two-hybrid assay to detect the interaction between She3 and She2 mutants at T109. Interaction between She3 and wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) was quantified by measuring β-galactosidase activity (N = 3). (**) P < 0.01. (C) Yeast two-hybrid assay to detect the interaction between Srp1 and She2 mutants at T109. Interaction between Srp1 and wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) was quantified by measuring β-galactosidase activity (N = 3). (****) P < 0.001.
The effect of phospho-null or phosphomimetic mutations at T109 on She3 and Srp1 binding was also tested in a yeast two-hybrid assay, as previously described (Shen et al. 2009). As shown in Figure 3B, the phosphomimetic T109D mutation decreased the formation of the She2–She3 complex twofold, while the T109A mutant had no impact on She2 interaction with She3. Another key interactor of She2 is the importin-α Srp1, which mediates the nuclear import of She2 via its interaction with an NLS at its carboxyl terminus (Shen et al. 2009). In a yeast two-hybrid assay to detect the interaction between She2 and Srp1 (Shen et al. 2009), a sixfold decrease in the interaction between She2 T109D and Srp1 was observed compared with She2 WT or She2 T109A (Fig. 3C). Altogether, these results show that phosphorylation at T109 inhibits She2 dimerization and its capacity to interact with its protein cofactors.
Phosphomimetic mutation at Threonine 109 inhibits She2 interaction with ASH1 mRNA and localization of this transcript
A recent structural study of She2 bound to the ASH1 mRNA localization element E3 revealed that the tetrameric structure of She2 is essential for binding RNA (Edelmann et al. 2017). Therefore, disruption of She2 dimerization is expected to affect its capacity to bind RNA. The impact of phospho-null or phosphomimetic mutations at T109 on She2 interaction with the ASH1 mRNA was tested using coimmunoprecipitation and RT-qPCR. As shown in Figure 4A, a 40% reduction in ASH1 mRNA binding was observed with the T109A mutation. The T109D mutation had an even greater effect, with a reduction of 80% in ASH1 mRNA binding. These results lead us to investigate the impact of a phosphomimetic mutation at T109 on the capacity of She2 to promote mRNA localization at the bud tip. Therefore, single molecule RNA FISH (smFISH) was performed on ASH1 mRNA in strains expressing She2 WT, She2 T109A, or She2 T109D proteins. As shown in Figure 4B,C, the reduced interaction between She2 T109D and ASH1 mRNA results in the nearly complete delocalization of this transcript compared to cells expressing wild-type She2. The She2 T109A mutant displays a milder phenotype, with increased accumulation of ASH1 mRNA within the bud instead of the bud tip (Fig. 4B,C). This result explains the reduced asymmetric distribution of the Ash1 protein when She2 T109A was tested in the genetic assay (Fig. 2A). Altogether, these results show that phosphorylation at T109 inhibits the capacity of She2 to bind ASH1 mRNA and to promote its localization at the bud tip. The hydroxyl group of T109 may participate in important hydrogen bonds between She2 monomers, which would explain the phenotype observed with the T109A mutant.
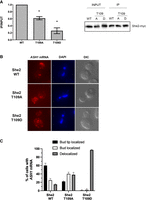
Mutation at T109 disrupts She2 interaction with ASH1 mRNA and its localization at the bud tip. (A) RNA immunoprecipitation of ASH1 mRNA by She2 wild-type or mutants. Myc-tagged wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) were immunoprecipitated using anti-myc antibody, followed by RNA purification and RT-qPCR analysis. (Left panel) Enrichment of ASH1 mRNA following immunoprecipitation is reported as a ratio of immunoprecipitate versus input (IP/INPUT), with wild-type She2 set as 1.0. (Right panel) Western blot of Myc-tagged wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) from input or immunoprecipitate (IP). (N = 3). (*) P < 0.05. (B) Fluorescence in situ hybridization (FISH) on ASH1 mRNA in yeast cells expressing wild-type She2 (WT), She2 T109A (T109A), or She2 T109D (T109D) proteins. (C) Quantification of ASH1 mRNA localization phenotypes in yeast cells expressing wild-type She2 (WT), She2 T109A, or She2 T109D proteins. N = 100 cells per strain, from two independent experiments.
Conclusion
Overall, this work shows that She2 is phosphorylated in vivo and that phosphorylation controls the function of this protein. Previous biophysical and structural studies on recombinant She2 have revealed that this protein assembles as a tetramer at physiological concentration (Müller et al. 2009). This tetrameric structure is essential for the interaction of She2 with both RNA and She3 (Edelmann et al. 2017). Recent observations from in-depth phosphoproteomic analyses in budding yeast revealed that phosphorylation at protein–protein interfaces constitutes an important regulatory mechanism (Lanz et al. 2021). Our data support these observations and show that phosphorylation at the interfaces controls the assembly of the She2 tetramer and constitutes an important regulatory mechanism for activity in vivo. When and where this phosphorylation occurs in the cell remains unclear.
We found that Threonine 109 is the most frequently phosphorylated residue in She2. A functional analysis of the phosphomimetic mutation at T109 revealed that this mutant strongly inhibits She2–She2 interaction, including heterologous interaction between She2 WT and She2 T109D, suggesting that this mutant is monomeric. Consequently, with its reduced oligomerization, the T109D She2 mutant displays strongly reduced interactions with both She3 and ASH1 mRNA, leading to the delocalization of this transcript. The T109D mutation also disrupted the interaction between She2 and the importin-α Srp1, which is surprising since our previous results showed that a monomeric She2 interacts more strongly with Srp1 compared to the She2 tetramer (Shen et al. 2009). In light of our new results, this model needs to be revisited and should take into account the role of phosphorylation at the dimerization interface of She2 in regulating the interaction between She2 monomers and Srp1.
Since the T109D mutant has such a negative effect on the function of She2, what could be the biological function of phosphorylation at this residue? The observation that the T109D mutation results in decreased She2 expression raises the possibility that T109 phosphorylation may lead to the destabilization and degradation of the protein. Indeed, phosphorylation at threonine residues in the middle of an α-helix, such as T109, promotes its destabilization and may impact the global structure and stability of a protein (Elbaum and Zondlo 2014). Since the She2 expression level is regulated by ubiquitin-targeting (Ziv et al. 2011), it will be interesting to explore the link between She2 phosphorylation and its ubiquitination.
Besides She2, other components of the budding yeast mRNA localization machinery are regulated by post-translational modifications. The locasome partner She3 is negatively regulated by phosphorylation at the S343, S348, and S361 residues, which inhibit its capacity to bind RNA but still maintain its interaction with She2 and Myo4 (Landers et al. 2009). These phosphorylation events have been linked to the stress response, such as osmotic stress or exposure to the reducing agent dithiothreitol (DTT) (Soufi et al. 2009; MacGilvray et al. 2020). This suggests that, in the presence of stress, the mRNA localization pathway may be inhibited, in part via She3 phosphorylation. Some of the phosphorylation events on She2 could play a similar role and may be part of a mechanism that inhibits the mRNA localization pathway in specific conditions.
MATERIALS AND METHODS
Yeast strain and DNA manipulation
Yeast growth was performed in YPD or synthetic selective media at 30°C. PCR-based gene disruption was performed as described previously (Guldener et al. 1996). All gene disruptions were confirmed by PCR of genomic DNA. To generate the strain with the GAL1prom-GST-SHE2 gene, the GAL1prom-GST cassette was generated by PCR from the plasmid pFA6a-KanMX6-PGAL1-GST (Longtine et al. 1998) and integrated in-frame of the SHE2 ORF in strain YS1052. The strains used are listed in Supplemental Table 5.
Plasmids construction
To generate She2 mutants, YCP22-She2-myc was PCR amplified using primers containing the appropriate mutation and cloned into PstI/KpnI sites of YCPlac22. The She2 mutants T109A and T109D were also cloned into pGADT7 and pGBKT7 plasmids for the yeast two-hybrid assay. All constructions were confirmed by Sanger sequencing. The plasmids used are listed in Supplemental Table 4.
Pro-Q Diamond phosphoprotein enrichment columns
Yeast strain K699 she2 + YCP22-SHE2-myc was grown in 500 mL of selection media until OD600 of 0.8. After centrifugation to pellet the cells, the pellet was resuspended in 5 mL of ice-cold lysis buffer (50 mM HEPES pH 7.5, 10% glycerol, 150 mM NaCl, 0.1% NP-40, 1 µM okadaic acid, protease inhibitors and RNAsin) and incubated for 30 min on ice. Using tissue lyser II with precooled adapter set and glass beads, cells were lysed for 2 min at 20 Hz, and centrifuged at 14,000g for 30 min at 4°C. The supernatant was collected and the protein concentration was measured with Bradford assay. The lysate was diluted with the wash buffer provided with the Pro-Q Diamond phosphoprotein enrichment kit (Molecular Probes # P33358, Thermo Fisher Scientific), to a final concentration of 0.1 mg/mL.
Pro-Q columns were prepared according to the manufacturer's instructions and equilibrated with 2× 1 mL of wash buffer. The diluted lysate was applied on the column, 1 mL at a time, followed by three washes with 1 mL of wash buffer. Flow-through and the three washes were conserved for western blotting. Elution of the phosphoproteins from the column was performed with 5× 250 μL of elution buffer (provided with the kit). Eluates were pooled and concentrated to a volume of 50 μL with 25 mM Tris pH 7.5, 0.25% CHAPS using the kit Vivaspin columns.
For phosphatase treatment, 10 μL of fastAP Thermosensitive Alkaline phosphatase (1 U/µL) was added to a solution of 0.1 mg/mL of yeast protein extract in 1× reaction buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 100 mM KCl, 0.02% Triton X-100, and 0.1 mg/mL BSA). The extract was incubated at 37°C for 3 h, before loading on the Pro-Q column.
For urea-mediated protein denaturation, the yeast lysate was diluted 10 times with the Pro-Q Diamond phosphoprotein Enrichment Kit Wash buffer containing 8M urea and run through the column. The column was washed three times with the same wash buffer containing 8M urea, before elution with the elution buffer.
She2 purification and mass spectrometry analysis
Yeast strain YS1052 GST-SHE2 with GAL1prom-GST-SHE2 was grown in YEP + 2% raffinose until OD600 0.2, where GST-She2 expression was induced with 3% galactose. At OD600 0.8, total proteins were extracted using glass beads in lysis buffer (20 mM HEPES pH. 7.5, 20% glycerol, 250 mM NaCl, 0.05% NP-40, 50× protease inhibitors + PMSF, 5 mM Na3VO4, and 25 mM Na3F). GST-She2 was purified using glutathione beads and eluted with 25 mM glutathione in 1× PBS. Following elution from the beads, GST-She2 was further purified on SDS–PAGE gel, stained with Coomassie blue and GST-She2 bands were cut from the gel.
Destaining of the gel was performed in 50% MeOH. The bands were shrunk in 50% ACN, reconstituted in 50 mM ammonium bicarbonate with 10 mM TCEP and vortexed for 1 h at 37°C. Chloroacetamide was added for alkylation to a final concentration of 55 mM. Samples were vortexed for another hour at 37°C. An amount of 1 µg of trypsin was added and the digestion was performed for 8 h at 37°C. Peptide extraction was conducted with 90% ACN. Extracted peptide samples were dried and solubilized in ACN 5% formic acid (FA) 0.2%. Samples were loaded on a homemade C18 precolumn (0.3 mm i.d. × 5 mm) connected directly to the switching valve and separated on a homemade reversed-phase column (150 µm i.d. × 150 mm) with a 56-min gradient from 10%–30% acetonitrile (0.2% FA) and a 600 nL/min flow rate on an Ultimate 3000 nano-LC (Dionex) connected to a Q-Exactive Plus (Thermo Fisher Scientific). Each full MS spectrum acquired with a 70,000 resolution was followed by 12 MS/MS spectra, where the 12 most abundant multiply charged ions were selected for MS/MS sequencing. Tandem MS experiments were performed using HCD at a collision energy of 25%.
The data were processed using PEAKS 7.0 (Bio-informatics Solutions) and the Saccharomyces cerevisiae UniProt database. Tolerances on precursors and fragments were 10 ppm and 0.01 kDa, respectively. Variable selected post-translational modifications were carbamidomethyl (C), oxidation (M), deamidation (NQ), phosphorylation (STY). Identification of phosphopeptides was further validated using Mascot. Cutoff for phosphopeptide identification probability was established at 70%. Three independent mass spectrometry analyses were performed, with peptide coverage of She2 of 76%, 86%, and 99%, respectively.
Yeast two-hybrid assays
Yeast two-hybrid assay was performed as described previously (Long et al. 2000; Shen et al. 2009). Plasmids pGADT7 or pGBKT7 expressing She2, She2T109A, and She2T109D were transformed in pJ69-4A she2Δ strain with the appropriate bait. Expression of the fusion proteins was confirmed by western blot. β-galactosidase activity was measured in solution using ONPG (o-nitrophenyl-D-galactopyranoside) as previously described (Shen et al. 2009), from at least three independent yeast cultures.
RNA immunoprecipitation and RT-qPCR analysis
Yeast cells were grown at 30°C in 50 mL culture of −TRP + 2% glucose to an OD600 ∼1.0. Cells were harvested by centrifugation and resuspended in 900 µL of lysis buffer (25 mM HEPES pH 7.5, 2 mM MgCl2, 0.1% IGEPAL, 150 mM KCl, 1 mM DTT, protease inhibitors, 2 mM VRC, and 100 U/mL of RNAsin). The cells were broken with glass beads by vortexing 6× 1 min, with 1 min pause on ice between each vortex. The lysate was centrifuged 10 min at 6500 RPM for 10 min, and the supernatant was recovered. From the supernatant, 600 µL was used for immunoprecipitation and 300 µL was kept as input. Immunoprecipitation of She2-myc was performed using 10 µg of 9E10 mouse anti-myc antibody added to the supernatant, and incubated overnight at 4°C. The day after, the solution was incubated for 4 h with 30 µL of Protein G Sepharose at 4°C. The beads were washed once with the lysis buffer for 5 min, followed by three washes of 5 min each with the wash buffer (25 mM HEPES pH 7.5, 150 mM KCl, 2 mM MgCl2). Elution was performed by incubation of the beads with 200 µL of 20 mM Tris pH 8.0, 100 mM NaCl, 2 mM EDTA, 5% SDS for 10 min at 65°C. From the eluate, 35 µL was used for western blot to detect She2-myc.
With the remaining volume of eluate, phenol/chloroform and ethanol precipitation was performed to recover the RNA that was immunoprecipitated with She2-myc. Reverse transcription was performed using half of the purified RNA with 0.2 µg of pd(N)6 oligos, incubated at 70°C for 5 min and quickly chilled on ice. A negative control without pd(N)6 oligos was prepared with the remaining RNA. Quantitative real-time PCR on cDNA was performed as described previously (Shahbabian et al. 2014), using primers for ASH1 and ACT1 (negative control). Cycle thresholds (Ct) for each triplicate of sample and input were averaged and immunoprecipitation enrichment was calculated by dividing the amount of IP over input using 2−ΔCT formula. Quantifications of mRNA levels of She2-myc WT, T109A, and T109D expressed from the YCP22 plasmid were performed by quantitative reverse transcription PCR on total RNA. Relative expression of SHE2 mRNA was calculated with both ALG9 and TAF10 reference genes. The primer sequences are available upon request.
Fluorescence in situ hybridization of ASH1 mRNA
Yeast strains K699 she2 + C3319 expressing YCP22-SHE2 WT, T109A, or T109D were grown in 50 mL selection medium until OD600 0.6–0.8. Cells were fixed for 45 min with 4% paraformaldehyde and harvested by centrifugation at 2500g for 4 min at 4°C. The cell pellets were washed thrice using ice-cold 1× buffer B (1.2 M Sorbitol, 0.1 M potassium phosphate at pH 7.5), with 4 min spinning at 2500g at 4°C between each wash. The pellets were resuspended in 1 mL of buffer B containing 20 mM vanadyl ribonucleoside (VRC), 28 mM β-mercaptoethanol, 0.06 mg/mL phenylmethylsulfonyl fluoride (PMSF) and 120 U/mL of RNAse inhibitor. Cells were transferred into a tube containing 250 units of lyticase (Sigma-Aldrich) and incubated for 10–12 min at 30°C until the cell wall was fully digested. The spheroplasts were pelleted by centrifugation for 4 min at 2500g at 4°C, and washed with 1 mL of ice-cold 1× buffer B. The pellets were resuspended in 750 µL of 1× buffer B, and 100 µL of spheroplast suspension was spotted per poly-L-lysine coated coverslip. The coverslips were stored at 4°C for 30 min. The spheroplasts were dehydrated by adding 5 mL of 70% ethanol and incubated for at least 20 min at −20°C before performing the in situ hybridization.
A set of 18 ASH1 smiFISH probes was designed using Oligostan R (Tsanov et al. 2016), and diluted to a concentration of 0.833 µM in Tris-EDTA pH 8.0 (TE) buffer. The diluted probe-set was hybridized with 50 µM of a Cy3-labeled FLAP oligo in a PCR machine. Coverslips were rinsed once with 1× PBS and incubated in 15% formamide freshly prepared in 1× SSC for 15 min at room temperature. The hybridization Mix 1 (2× SSC, 34 µg of E. coli tRNA, 30% formamide, and FLAP-bound ASH1 probe-set) and Mix 2 (20 µg of RNAse-free BSA, 4 mM VRC, and 21% dextran sulphate) were put together and spotted on the coverslips. The coverslips were incubated in an airtight hybridization chamber at 37°C overnight (Querido et al. 2020). The coverslips were washed twice for 30 min with freshly prepared 15% formamide/1× SSC solution at 37°C and rinsed twice in 1× PBS before mounting. The coverslips were placed on slides with Vectashield antifade mounting medium containing DAPI (VECTOR Laboratories). Imaging was performed using a Zeiss Axio-Imager Z2 upright microscope.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank the Proteomic Core facilities at the Institute for Research on Immunology and Cancer (Université de Montréal) and the Montreal Clinical Research Institute (IRCM). P.C. holds a research chair from Fonds de Recherche du Québec-Santé (FRQS). This project was funded by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2019-05023) to P.C.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079555.122.
-
Freely available online through the RNA Open Access option.
- Received December 14, 2022.
- Accepted February 24, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Nastaran Farajzadeh is joint first author of this paper “Phosphorylation controls the oligomeric state of She2 and mRNA localization in yeast.” Nastaran was a PhD candidate in the laboratory of Dr. Pascal Chartrand at Université de Montréal, where she investigated the mechanisms regulating mRNA localization in budding yeast. She has recently taken a postdoctoral position at the Sainte-Justine Hospital Research Center in Montréal.
What are the major results described in your paper and how do they impact this branch of the field?
In the present study, we determined that She2, the key RNA-binding protein involved in mRNA localization at the bud tip of yeast cells, is phosphorylated at several residues. We also uncovered that phosphorylation at residues located at the She2 dimerization interface impacts She2 oligomerization, interaction with its cofactors, and binding to mRNA. These findings suggest that phosphorylation regulates She2 oligomerization, which emerges as a key regulatory step in mRNA localization at the bud tip. Since She2 plays a key role in mRNA localization, the present study will help us better understand how mRNA localization machinery is regulated in yeast and other organisms.
What led you to study RNA or this aspect of RNA science?
So far, most studies have focused on the roles of post-translational modifications in the translational regulation of localized mRNAs. However, how post-translational regulation may affect the mRNA localization machinery itself is not clear. While She2 is the key RNA-binding protein that plays a critical role in mRNA localization at the bud tip, its regulation was still poorly understood, which led us to study the role of its phosphorylation on the mRNA localization.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Throughout my childhood, I was fascinated by books, which helped in cultivating my curiosity. Fortunately, I had an excellent reading resource in our home, my father's library! Several books attracted my attention. I was a big fan of Jules Verne and Isaac Asimov. A Trip to the Moon by Jules Verne fascinated me and I read it several times. Reading books during my childhood sparked my curiosity, which helped in driving me to pursue science later, where I could find answers to my many questions.
If you were able to give one piece of advice to your younger self, what would that be?
Due to the nature of scientific study, persistence and perseverance are necessary. Given that many experiments are challenging to conduct or completely unsuccessful, persistence is essential to find a solution for the failed experiment and overcome the difficulties in this journey.
What were the strongest aspects of your collaboration as co-first authors?
Dr. Karen Shahbabian and I collaborated on this publication to accelerate the research process and increase our team productivity. Throughout my research studies, I learned how important it is for scientists to be able to work together, share ideas, and build on one another's research.








