
Direct dsRNA preparation by promoter-free RCT and RNase H cleavage using one circular dsDNA template with a mismatched bubble
- 1College of Food Science and Engineering, Ocean University of China, Qingdao 266550, Shandong, China
- 2Laboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266235, Shandong, China
- 3Department of Biochemistry and Molecular Biology School of Basic Medicine, Qingdao University, Qingdao 266071, Shandong, China
- Corresponding authors: liangxg{at}ouc.edu.cn, ar{at}ouc.edu.cn
-
↵4 These authors contributed equally to this work.
Abstract
Double-stranded RNA (dsRNA) has aroused widespread interest due to its effects on immunity and applications based on RNAi. However, the in vitro preparation of dsRNA is costly and laborious. In this study, we have developed a novel and interesting method designated as pfRCT (promoter-free rolling-circle transcription) for direct, facile, and efficient dsRNA preparation. This method generates equal amounts of sense and antisense strands simultaneously from a single circular dsDNA template. To initiate transcription by T7 RNA polymerase without directional preference, a 9–15-bp bubble (mismatched duplex with strong sequence symmetry) is introduced into the template. During RCT, all the necessary reagents, including the template, NTPs, RNA polymerase, RNase H, and Helpers, are present in one pot; and the just-transcribed RNA is immediately truncated by RNase H to monomers with the desired size. The ends of the dsRNA product can also be simply sealed by T4 RNA ligase 1 after pfRCT. This new approach is expected to promote the applications of dsRNA.
Keywords
INTRODUCTION
Efficient preparation of double-stranded RNA (dsRNA) is important to meet the requirements of both scientific research and industry. With the soaring development of RNA interference (RNAi), the demand for dsRNA which can be digested to small interfering RNA (siRNA) will increase significantly in fields including agriculture, aquaculture, and the medical industry (Posiri et al. 2013; Yu et al. 2013; San Miguel and Scott 2016; Chaimongkon et al. 2020; Parenrengi et al. 2020). In addition, dsRNA structures exist ubiquitously in nature, such as in rRNA, mRNA, pre-miRNA, viroids, and dsRNA viruses (Seidl et al. 2013). If it is easily available, various new researches can be expected. However, traditional solid-phase synthesis usually yields low outputs and has limitations on RNA length within ∼100 bp (Cai et al. 1998; Gilar 2001; Hogrefe et al. 2013; Wang et al. 2018). Purification of dsRNA from cells for in vivo preparation is cumbersome (Chalupnikova et al. 2013; Posiri et al. 2013; Chaimongkon et al. 2020; Parenrengi et al. 2020; Hashiro et al. 2021). The linear transcription strategy can produce a sufficient quantity of dsRNA, but the products attach a 5′-triphosphate group, which may induce an immune response (Kim et al. 2004). Additionally, RNA polymerase (RNAP) has to carry out initiation for each turnover in linear transcription (Donzé and Picard 2002; Sohail et al. 2003; Hameed et al. 2017).
Rolling-circle transcription (RCT) shows great potential for dsRNA production. RCT can synthesize a long RNA product with a sequence repeated dozens to hundreds of times after each initiation. After the long RNA is truncated by RNase H, the products attach a 5′-phosphate, similar to the 5′ end of siRNA. However, RCT has its own problems. For example, the promoter sequence is also transcribed and involved in the product. In addition, another RNAP can bind again to the promoter on the DNA template after initiation and block the elongation by the first RNAP. In the case of dsRNA preparation using a long dumbbell-like or Y-form circular ssDNA template (Seyhan et al. 2006; Jang et al. 2018), RCT becomes difficult because RNAP has weak strand displacement ability. In the case of RCT synthesizing two complementary ssRNA strands separately, the procedures are tedious because two templates are needed, and the ratio is not so easily controlled to be 1:1 (Han et al. 2017; Kim et al. 2017). Furthermore, during the synthesis of each ssRNA, undesired by-products are produced in some cases (Daubendiek et al. 1995; Ohmichi et al. 2002; Seidl et al. 2013; Ducani et al. 2014).
In 1994, Aiyar et al. (1994) showed that a linear dsDNA template without a promoter sequence (promoter-free) but containing a 12-bp bubble could be transcribed by Escherichia coli RNAP in both directions. At the bubble, all nucleotides are mismatched and in a dissociated (open) state. Inspired by this, we developed a novel strategy to directly synthesize dsRNA from only one promoter-free circular dsDNA template containing a 9–15-bp bubble (mismatched duplex) (Fig. 1), which is designated as pfRCT (promoter-free Rolling-Circle Transcription). T7 RNAP can initiate transcription without directional preference so that 50% of dsDNA template molecules are transcribed to produce sense strands, and the other to produce antisense strands (ASs). The transcripts of long RNA can be site-specifically cleaved by RNase H quickly so that the desired dsRNA is produced continuously and efficiently (Drolet et al. 1995; Hraiky et al. 2000; Amon and Koshland 2016; Chedin and Benham 2020). More interestingly, the free ssRNA ends of dsRNA products can be sealed further to improve their nuclease resistance (Fig. 1A). This approach is promising to promote the applications of RNAi and helpful for clarification of the transcription mechanism in vivo (Jin et al. 2014; Nellimarla and Mossman 2014; Allen et al. 2017; Paulsen et al. 2019).
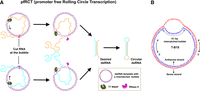
Outline of the pfRCT strategy for preparation of dsRNA. (A) Schematic illustration of pfRCT using the combination of RCT and RNase H cleavage. T7 RNAP initiates transcription without direction preference at the bubble. During transcription, the RNA products bind to the ssDNA parts at the bubble to form short RNA/DNA duplexes for RNase H cleavage (Drolet et al. 1995; Hraiky et al. 2000; Amon and Koshland 2016; Chedin and Benham 2020). The synthesized two complementary RNA strands hybridize to form the desired dsRNA. RNA cleavage can be accelerated by using short DNA Helpers, which hybridize with RNA products transcribed from the bubble. The obtained dsRNA can be circularized by T4 RNA ligase 1 (T4 Rnl1). (B) An example of sequence design. The 109-bp dsDNA template (T-B15) without promoter sequence contains a 15-bp bubble.
RESULTS
Construction of the circular dsDNA template involving a bubble and efficient dsRNA synthesis using pfRCT
The 109-bp circular dsDNA template involving a 15-bp bubble (Fig. 1B) was constructed using a novel method shown in Figure 2A. After hybridization of two ssDNA fragments with the prepared circular ssDNA (C-AS) (see Supplemental Fig. S2 for details) to form a dsDNA structure with a bubble and two nicks, ligation was carried out to seal the nicks (Fig. 2A). As shown in Figure 2B (lanes 4 and 6), the new bands with lower mobility indicate that the two fragments (S-a and S-b) hybridized with C-AS (prepared from two fragments of As-a and As-b). After ligation using either T4 DNA ligase at 37°C or Taq DNA ligase at 65°C, bands with even lower mobility are observed (lanes 8 and 10). After digestion by Exonuclease I and Exonuclease III to remove linear by-products and substrates (lanes 9 and 11), the left bands are assigned as the circular dsDNA template (T-B15) with a bubble. Interestingly, two bands are left, which can be assigned as topological isomers of the circular dsDNA with a different linking number (Lk). For this 109-bp circular dsDNA, the largest Lk should be 10 (10.5 bp/turn for B-DNA). Usually, larger Lk gives higher mobility due to the tight structure (Liang et al. 2006). These two bands obtained after ligation at 37°C are assigned as Lk9 and Lk10 (lane 9), and those obtained at 65°C as Lk8 and Lk9 (lane 11). For Lk8 and Lk9, the dissociated parts during ligation are ∼20–25 and 10–15 bp, respectively (as shown in Supplemental Fig. S3C). It is reasonable because several base pairs flanking the bubble prefer to dissociate at 65°C (Liang et al. 2006).
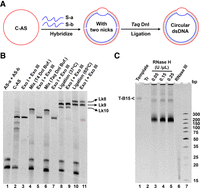
Construction of a circular dsDNA template involving a bubble of mismatched duplex and the transcription results. (A) Construction strategy. After hybridization, the nicks are sealed by Taq DNA ligase. The topology of prepared circular dsDNA (T-B15) is controlled by changing the ligation temperature. (B) dPAGE analysis (8%, 8 M urea) of ligation. Lane 1, the short ssDNA AS-a and AS-b to construct C-AS. Lane 2: the circular ssDNA (C-AS). Lanes 4 and 6: the hybridization between C-AS and two complementary short ssDNA (S-a and S-b). The products were hybridized in the T4 Dnl buffer (lane 4) and Taq Dnl buffer (lane 6), respectively. Lanes 8 and 10: products obtained by sealing nicks with T4 Dnl (37°C) and Taq Dnl (65°C), respectively. Lanes 3,5,7,9, and 11: digestion of the products in lanes 2,4,6,8, and 10 using Exonuclease I and Exonuclease III. (C) dPAGE analysis (8%, 8 M urea) of transcription. Lane 1: T-B15; lane 2: conventional RCT (without RNase H). “Tr” refers to “Transcription”; lanes 3–5: pfRCT at various concentrations of RNase H; lane 6: the product in lane 5 was treated by ShortCut RNase III (digest dsRNA to short dsRNA fragments of 18–25 bp); lane 7: Ultralow range DNA Ladder (ULR). Transcription conditions: [T-B15 (prepared in 65°C)] = 50 nM, [T7 RNAP] = 2 U/µL, [RNase Inhibitor] = 2 U/µL, [NTP]each = 2 mM, 1× transcription buffer, 37°C for 4 h. For pfRCT, RNase H was present.
The transcription results using T-B15 as the template (obtained by Taq ligase ligation) are shown in Figure 2C. In the absence of RNase H, only a weak band is observed in the gel well (lane 2), indicating that long RNA with tandem repetitive sequences was produced with low efficiency. However, in the presence of RNase H during transcription, strong bands appear at the position of ∼200 bp (DNA marker) as well as in the well (lanes 3–5), demonstrating that the transcription efficiency was greatly improved with the aid of RNase H digestion. Obviously, the transcribed RNA can bind to the template at the bubble to form an RNA/DNA duplex so that RNase H can cleave the RNA strand (Fig. 1A; Supplemental Fig. S1). Once the cleavage by RNase H occurs, the length of RNA complementary to the bubble becomes short (6–9 nt) and the cleaved RNA is easy to dissociate. In addition, the transcription of the next cycle can help the cleaved RNA dissociate from the DNA template. Accordingly, the expected length of produced dsRNA is 94 bp, attaching four 6–9-nt-long ssRNA tails at the ends. This may be the reason that the bands appear close to the 200-bp DNA marker. After digestion of the transcript shown in lane 5 by ShortCut RNase III (a dsRNA digestion enzyme, as the Dicer in vivo), smeared bands with higher mobility (∼15–25 bp) appeared (lane 6), demonstrating that the transcription products are dsRNA (see also Supplemental Fig. S4B). Obviously, pfRCT for efficient dsRNA preparation is realized due to the continuous transcription and removal of RNA products from the template immediately (see the proposed mechanism in Supplemental Fig. S1).
When T4 DNA ligase was used instead of Taq DNA ligase, the transcription efficiency decreased greatly (Supplemental Fig. S3B). We have shown that higher ligation temperatures prefer to form DNA catenanes with smaller Lk (Liang et al. 2006). Accordingly, we believe that the topoisomers of T-B15 with a smaller Lk (Lk8) may be favorable for pfRCT. If the Lk is larger (e.g., Lk9), the initiation may occur more easily, but the cleavage by RNase H should become difficult. In the case of Lk8, R-loop prefers to form at the bubble during RCT so that RNase H binds and cuts RNA easily. When the RNA products obtained by RCT without RNase H (lane 2, Fig. 2C) were subsequently digested by RNase H, most transcripts still stayed in the gel well, indicating that once the long dsRNA forms, it is hard to be cleaved by RNase H (lanes 3 and 4, Supplemental Fig. S4A). One possible reason is that the bubble in the dsDNA template hybridizes with difficulty with the bubble in long dsRNA due to topological constraints. If the time is long enough, all long dsRNA can be truncated (data not shown).
When inorganic pyrophosphatase (IPP) was added (hydrolyzing pyrophosphate to release Mg2+) (Cunningham and Ofengand 1990), the yield (calculated by the NTP consumption) increased from 17.5% to 57.5% (50 nM T-B15 was used). The effects of NTP concentration and template concentration were also investigated, and the optimal conditions are 50 nM DNA template, 2 mM NTP (each), and 1 U/mL IPP (Fig. 3). The effects of the size (5–15 bp) and sequence of the bubble on transcription and RNase H truncation efficiency were also investigated, and the 15-bp-long bubble was found most suitable (Fig. 4). For some special sequences such as PolyA and PolyT, almost no transcript was observed.
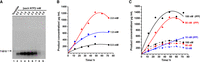
Effects of NTP concentration, dsDNA template concentration, and the addition of IPP on pfRCT. (A) NTP concentration. Other conditions: [T-B15] = 50 nM, without IPP, 24 h, 37°C. Analyzed by 1% agarose. (B) Time courses under various concentrations of each NTP. Other conditions: [T-B15] = 50 nM, with 1 U/mL IPP. (C) Time courses under various template concentrations, with or without IPP. [NTP]each = 2 mM, [IPP] = 0 or 1 U/mL. The reaction conditions are shown in Materials and Methods.
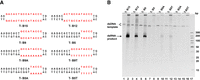
Bubble size and sequence greatly affect the pfRCT efficiency. (A) Sequence information for bubbles (red letters). The direction of the upper sequence from left to right is 5′ → 3′. Other regions of the template are the same with T-B15 shown in Figure 1B. (B) Results of pfRCT. Samples were visualized on an 8% denaturing PAGE (8 M urea). Lanes 1,3,5,7,9,11,13, and 15: dsDNA templates only; lanes 2,4,6,8,10,12,14, and 16: pfRCT results; lane 17: marker of dsDNA.
Evidence for proving the products are dsRNA and RNase H truncates the transcripts at the bubble site
As shown previously, when RCT was carried out in the absence of RNase H, only long RNA products were produced (lane 2, Fig. 2C). When these RNA products were subsequently cleaved by RNase H with the aid of one or a pair of ssDNAs (Helpers), which have the same sequence as the bubble on the dsDNA template (Fig. 5A), the RNA products can be truncated to shorter ones (lanes 5–7, Fig. 5B). However, when the ssDNA was partially complementary to the duplex part of T-B15, no digestion was observed (lanes 8–9). These results demonstrate that the long RCT products are dsRNA with mismatched bubbles, which can form RNA/DNA duplexes with the ssDNA Helpers. When a 118-bp circular dsDNA with the T7 promoter sequence (no mismatch) was used as the template for transcription in the presence of RNase H, no short RNA monomer was observed (lane 5 in Fig. 5C). In this case, only the AS can be transcribed, and ssRNA is produced. Accordingly, the mismatched bubble on T-B15 is necessary to cause RNA cleavage by RNase H during pfRCT.
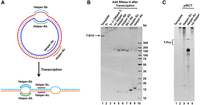
Confirmation of the structure of pfRCT product via digestion by RNase H with the help of various Helpers. (A) Illustration of the position of various Helpers. “Helper-Ab” and “Helper-Sb” have the same sequences with the bubble in “T-B15.” “Helper-Ac” and “Helper-Sc” have the same sequences with a part of the duplex in “T-B15” (sequence information and length are shown in Supplemental Table S1). (B) The digestion products were analyzed by 10% PAGE. Lane 1: T-B15 template; lane 2: RCT product without RNase H; lane 3: product in lane 2 was treated by DNase I (to remove T-B15); lane 4: product in lane 3 was treated by RNase H without any Helper; lanes 5–9: product in lane 3 was digested by RNase H with the help of various Helpers; lane 10: size marker. The reaction conditions are shown in Materials and Methods. (C) Transcription products (8% dPAGE with 8 M urea) of “T-pro” (a 118-bp circular dsDNA template with promoter sequence). Lane 1: T-Pro template; lane 2: products obtained by RCT; lanes 3–5, products obtained by pfRCT with/without the aid of Helper (1/0 µM). The reaction conditions are shown in Materials and Methods.
To prove that the short RNA products are dsRNA, eight 25-nt ssDNAs (SDs) complementary to various positions of possible RNA products were designed (Supplemental Fig. S6A). After pfRCT, these SDs were used for RNase H digestion (Supplemental Fig. S6B). For all combinations, almost no mobility change of pfRCT products was observed, demonstrating that they are dsRNA as expected. Even when the ssDNAs were complementary to the expected ssRNA ends (SD1–SD4), no obvious shortening occurred (lanes 4–5, 8–12, Supplemental Fig. S6B), probably because the ssRNA ends are too short for hybridization, especially after several nucleotides are removed by RNase H. Certainly, the ssRNA ends (6–9 nt) on the dsRNA can be removed by the nuclease of RNase T1 if necessary (Nwokeoji et al. 2017).
The addition of an ssDNA Helper for RNase H cleavage greatly prompts pfRCT using the dsDNA template involving a bubble with a completely symmetric sequence and fewer mismatches
Considering that RNase H binding to the hybrid formed between the ssRNA product and DNA template at the bubble may slow down elongation by RNAP, we modified the molecular design as shown in Figure 6. The main points are described as follows. (i) In the newly designed 15-bp bubble (T-B15S, Fig. 6A), only five mismatches are involved; (ii) to increase the symmetry, the bubble is designed to have a palindromic sequence; (iii) the truncation of the long dsRNA is carried out by using a short ssDNA Helper (Fig. 6B), the sequence and length of which is the same as the bubble. It is noteworthy that only one ssDNA Helper is required because the bubble sequence is palindromic. This Helper can form a palindromic duplex involving five mismatches, which is very unstable (Tm lower than 10°C). After the transcription is initiated, the less mismatches (as compared with T-B15) would make R-loop formation more difficult, because the RNA is more easily squeezed out by rehybridization of the dsDNA template at the bubble during RCT. However, the RNA products are more difficult cleaved by RNase H on the template. Accordingly, the truncation is designed to carry out with the Helper after the RNA dissociates from the template (Fig. 6B). Higher efficiency of pfRCT can be expected, because the interference between cleavage and elongation can be greatly reduced.
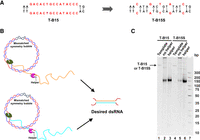
Efficient pfRCT by using a dsDNA template with fewer mismatches and symmetric sequences. (A) Sequence of bubbles. The newly designed bubble (only five mismatches) in T-B15S is completely symmetric (palindromic). (B) Schematic illustration of pfRCT with the aid of a DNA Helper to improve the cleavage efficiency of RNase H. For T-B15S, only one 15-nt ssDNA Helper is added. For T-B15, two 15-nt DNA oligonucleotides with the same sequences as the bubble are required. Once the ssRNA is transcribed, it hybridizes with the ssDNA Helper and is cleaved by RNase H. Accordingly, the transcription and truncation proceed simultaneously without interference with each other. (C) Electrophoresis analysis (10% PAGE) of pfRCT products in the absence or presence of ssDNA Helpers. Lanes 1 and 4: dsDNA templates; lanes 2 and 5: pfRCT for T-B15/T-B15S without Helper; lanes 3 and 6: pfRCT for T-B15/T-B15S with Helper. Other conditions: [Template] = 50 nM, [each NTP] = 2 mM; [each Helper] = 1 µM.
As shown in Figure 6C (lane 5), for T-B15S in the absence of Helper, most transcripts were blocked at the well (only a small portion ran to the middle region of the gel), indicating that the transcripts are mainly long RNAs. With the aid of the 15-nt-long ssDNA Helper, shorter RNA products were observed (lane 6). Obviously, the presence of the ssDNA Helper improved greatly the efficiency of pfRCT (see also Supplemental Fig. S7B). For T-B15, the main RNA products are shorter ones even in the absence of two ssDNA Helpers (lane 2), although the efficiency of pfRCT was also improved to some extent when Helpers were present (lane 3 in Fig. 6C; Supplemental Fig. S7C). No other bands or smearing of the bands were observed, indicating that the digestion was relatively site-specific even 15-nt-long ssDNAs were used. In addition, the opportunity for nonspecific cleavage by RNase H decreases greatly because the newly synthesized ssRNAs form the dsRNA immediately.
The effect of Helper length (8–15 nt) was also investigated (Supplemental Fig. S8). Obviously, the 15-nt-long Helper was most efficient, for which 1.0 µM was enough (more than 90% transcript was cleaved to the desired size). For the 12-nt-long Helper, only higher concentrations (e.g., 10 µM) could result in relatively complete cleavage. For Helpers of 10 or 8 nt, cleavage could not carry out efficiently even at 20 µM (Supplemental Fig. S8). It can be explained that shorter Helpers are difficult to hybridize with RNA at the reaction temperature of 37°C. It is noteworthy that the hybridization of short Helpers is also less efficient at lower temperatures due to the secondary structure of long RNA. Accordingly, efficient cleavage can only be obtained for the Helper of 15 nt in this case. Helpers longer than 15 nt usually lower the specificity of cleavage.
We also carried out pfRCT using various lengths of templates (Supplemental Figs. S9–S11). For the template of 59-bp-long T-CC59-15, which contains a 15-bp bubble (with continuous mismatches like T-B15), the band for RNA product was observed only in the gel well, indicating that long RNA with the tandem repetitive sequence was not truncated by RNase H (lanes 1–6 in Supplemental Fig. S9B). Similar results were obtained for 59-bp-long T-CC59-12 (with a 12-bp bubble, lanes 7–12 in Supplemental Fig. S9B) and T-CC59-15S (with a 15-bp bubble of palindromic sequences, Supplemental Fig. S10A). When the 15-nt-long Helper was used, short RNA products were obtained (lane 4 in Supplemental Fig. S10B), although the RNase H cleavage was not so efficient as that for T-B15S. For longer templates of 96 or 155 bp, efficient pfRCT was carried out even in the absence of a Helper (Supplemental Fig. S11). Accordingly, we conclude that pfRCT can be used as a general approach to efficiently synthesize at least 59–155-bp-long dsRNA. For longer products (e.g., >200 bp), transcription using linear dsDNA templates is more favorable (Supplemental Fig. S11), because preparation of a large-sized circular dsDNA with a mismatched bubble is challenging, and the superiority of RCT becomes less.
Unexpectedly, we found that the transcription prefers a little bit to occur in one direction even when the bubble has a palindromic mismatched sequence (e.g., T-B13S, Supplemental Fig. S12). The hybridization results show that transcription prefers to use the sense strand as the template (lanes 2–5 in Supplemental Fig. S12C, lanes 2–7 in Supplemental Fig. S10C). The possible reason is that the sequences of the dsDNA flanking the bubble may affect the preference of T7 RNAP's binding even when they are completely different from the T7 promoter. This was confirmed by using symmetric sequences (see designs in Supplemental Fig. S12), where almost no direction preference was observed (lanes 8, 9, 13, and 14 in Supplemental Fig. S12C). Accordingly, not only the sequence symmetry at the bubble but also the adjacent dsDNA sequences affect the direction preference of transcription.
Circularization by T4 Rnl1 of the dsRNA prepared by pfRCT
Considering that circular RNA is much stable compared with a linear one (Abe et al. 2007), we tried to circularize the dsRNA products prepared by pfRCT (Fig. 7A). As shown in Figures 1 and 6, these dsRNAs involve a special structure of double-stranded part in the middle and single-stranded tails at both ends (5′-phosphate and 3′-OH), which are ideal substrates for ligation by T4 Rnl1 (a ligase using ssRNA as the substrate). It has been reported that the minimum length requirement for circularization by T4 Rnl1 is 6–8 nt (Petkovic and Müller 2015; Müller and Appel 2017). As shown in Figure 7B, after ligation for 6 h, the mobility of the dsRNAs increased obviously (compare lanes 3 and 7 with lanes 2 and 6), indicating that the ligation was successfully carried out. In addition, after digestion by RNase R (an exonuclease digesting linear RNAs but not circular ones), the ligation products remained unchanged (see lanes 4 and 8), further demonstrating that they are circular RNA without free ends. It should be noted that the sealed RNA here has a dumbbell structure (Fig. 7A), distinct from normal circRNAs (circular RNAs).
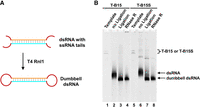
Circularization by T4 Rnl1 of the dsRNA obtained by pfRCT. (A) Schematic diagram of the ligation strategy. (B) Circularization results analyzed by dPAGE (8%, 8 M urea). Conditions for pfRCT: [Template] = 50 nM, [NTP]each = 2 mM, [T7 RNAP] = 2 U/µL, [RNase Inhibitor] = 2 U/µL, 1× T7 RNAP buffer, [RNase H] = 0.25 U/µL, [Helper] = 1 µM, 37°C for 4 h. Lanes 1 and 5: dsDNA templates; lanes 2 and 6: dsRNAs produced using pfRCT; lanes 3 and 7: the products in lanes 2 and 6 were ligated by T4 Rnl1; lanes 4 and 8: ligation products in lanes 3 and 7 were further digested using RNase R. Conditions for ligation: 50% (volume) of transcription solution, [T4 Rnl1] = 0.5 U/µL, 0.5× T4 Rnl1 buffer, [ATP] = 1 mM, [RNase Inhibitor] = 2 U/µL, 37°C, 6 h.
DISCUSSION
The method of pfRCT is facile and efficient for dsRNA preparation
The novel method for the preparation of dsRNA shown here can be simply carried out in a one-step way, that is, all the components are mixed, including a circular dsDNA template with a bubble, NTPs, RNAP, RNase H, and one or two DNA Helpers. The most difficult part is the design and preparation of circular dsDNA templates, especially for control of the linking number (Lk). Circular dsDNA templates of various lengths are prepared simply by hybridization of circular ssDNA with complementary linear strands, followed by ligation at various temperatures. Higher temperatures result in topoisomers with smaller Lk numbers, because the mismatched base pairs (at the bubble) cannot form a duplex at higher temperatures (Liang et al. 2006).
Our approach can be used for the preparation of dsRNA using a relatively low concentration of dsDNA templates, because the pfRCT can be carried out continuously with the removement of RNA products from the template immediately. In the case of preparing a circular dsRNA with a dumbbell structure, purification can be simply done by HPLC (high-performance liquid chromatography) or other chromatography approaches because only other small molecules are present after enzymatic removal of DNA (by DNase) and linear RNA (by exonuclease). There is no obvious limitation in sequence [only some extremely special sequences such as poly(A) and poly(T) should be avoided in the bubble], and the template design is not difficult. The bubble should be palindromic with a proper ratio of mismatches. When transcribing for 4 h, the yield of dsRNA can reach 2.25 µg in a 10 µL reaction system (calculated through the time course shown in Supplemental Fig. S7C), in which only 0.036 µg of the circular dsDNA template was used. Additionally, the cotranscription with two linear templates (containing a promoter sequence) was also performed under the same conditions. The yield of dsRNA was 0.836 µg, accompanied by the generation of significant by-products (Supplemental Fig. S13). Therefore, pfRCT not only has a higher yield than the cotranscription but also produces purer products. For some applications, such as RNAi for plants to kill viruses or pathogenic bacteria, the prepared dsRNA can be directly utilized without further purification (Posiri et al. 2013; Yu et al. 2013; San Miguel and Scott 2016; Chaimongkon et al. 2020; Parenrengi et al. 2020).
The mechanism of pfRCT, which can explain well the highly efficient transcription, was proposed (Supplemental Fig. S1). The smaller Lk makes the bubble in a completely open state so that RNAP can bind and initiate transcription. The bubble can form a mismatched duplex during elongation to cushion the stress caused by bubble formation, which is helpful for continuous elongation. On the other hand, the proceeding of polymerase will be impeded if the transcribed RNA hybridizes to the template strand at the bubble too tightly. Appropriately, RNase H can cleave the RNA strands in this DNA/RNA hybrid so that the smoothing transcription can be carried out. At other full-match positions, the RNA can be squeezed out quickly during elongation. For the small (<200 bp) circular dsDNA templates we use here, topological stress can transmit along the template easily so that no strong constraint occurs to slow down RCT. Accordingly, RNase H can only cleave the nascent RNA at the bubble site. This is the reason why site-specific cleavage is realized during pfRCT.
Because the newly produced RNA strands can be truncated in time, no extra long RNA (impeding the approach of NTPs to the DNA template) is attached to the template so that the elongation can proceed efficiently all the time. This is impossible for a circular ssDNA as the template for RCT, because almost all RNA hybridizing on the template can be cleaved. In addition, it is also impossible for a circular dsDNA template without a bubble (but only with a smaller Lk), because the two DNA Helpers targeting the same position are completely complementary. Furthermore, the circularization of dsRNA products makes pfRCT more applicable, because circular nucleic acids are much more stable in vivo (Abe et al. 2007). It should be noted that other factors of easy ligation by T4 Rnl1 should be considered during the design of the bubble sequence. For example, it has been reported that T4 Rnl1 exhibits certain terminal base preferences (3′-end: A > G ≥ C > U; 5′-end: pC > pU > pA > pG) (Petkovic and Müller 2015; Müller and Appel 2017). Both the binding position and length of DNA Helpers can be adjusted, because it can be simply done by changing the bubble sequence.
Reconsideration of the function of the promoter for transcription initiation
In this study, we show that T7 RNAP can easily initiate transcription in a promoter-free circular dsDNA template with smaller Lk values. Based on our results, we propose that the promoter should be redefined as a “locator,” whose main function is to determine the exact starting point and direction for transcription. Earlier studies by Tripatara and deHaseth (1993) and Aiyar et al. (1994) also reported that E. coli RNAP can initiate transcription from a mismatched bubble in a promoter-free dsDNA template.
For transcription initiation, the opening of dsDNA to form a bubble of sufficient size (at least 10 bp) is the basic requirement (Zuo et al. 2020). However, it is almost impossible to generate a bubble of such size in vivo without consuming enough energy, despite the preference of negative supercoiling to induce bubble formation (Brahms et al. 1985). Accordingly, for strict control of transcription, only after the RNAP binds tightly to the promoter, it can help open a bubble to initiate transcription. However, this tight binding often results in the generation of abortive transcripts during the early stages of transcription. Obviously, a mismatched bubble is much easier to initiate transcription without the stress to open a complementary duplex part, and the generation of abortive transcripts may be avoided (Zhou and Martin 2006; Zuo et al. 2020). The transcription goes easily to the elongation state, because RNAP binds weakly to the nonpromoter sequence.
Conclusion
For the highly efficient preparation of desired dsRNAs, a novel strategy called pfRCT has been constructed. By using a promoter-free circular dsDNA with a 15-bp bubble as the template, the bidirectional transcription can be carried out to produce dsRNA directly. The sequences of the bubble as well as its adjacent dsDNA should be designed with high symmetry, and the template should be prepared under high temperatures. The nascent ssRNA strands can be cleaved from the template in time by RNase H to avoid their negative effects on elongation. The tacit cooperation between T7 RNAP and RNase H greatly facilitates the production efficiency. Simply by mixing all the components [including T7 RNAP, RNase H, NTPs, dsDNA template, ssDNA Helper(s), and transcription buffer] together and incubating for a period, the desired dsRNA could be efficiently produced with high purity. By using our approach, more applications of RNAi become promising in agriculture, fishery, RNAi vaccine, and cancer immunotherapy. Our finding is also helpful for understanding the transcription mechanism. If the direction of transcription can also be controlled by this pfRCT strategy, production of ssRNA is also possible, and some demerits for linear transcription are promising to be avoided (Cazenave and Uhlenbeck 1994; Pleiss et al. 1998; Helm et al. 1999; Zaher and Unrau 2004; Sarcar and Miller 2018).
MATERIALS AND METHODS
Materials
All oligonucleotides were synthesized by Sangon Biological Engineering Technology, and the sequences are listed in Supplemental Table S1. For ligation, a phosphate was introduced to the 5′-position of DNA by using T4 polynucleotide kinase (T4 PNK). T4 DNA Ligase (T4 Dnl), Exonuclease I (Exo I), Exonuclease III (Exo III), T7 RNA polymerase (T7 RNAP), RiboLock RNase Inhibitor, NTPs, RNase free water, and DNA ladder were purchased from Thermo Scientific. RNase H, Taq DNA Ligase (Taq Dnl), IPP, T4 RNA Ligase 1 (T4 Rnl1, T4 Rnl), and ShortCut RNase III were provided by New England Biolabs. Ultra GelRed and Gene Green were from Vazyme. RNase R was obtained from GeneSeed (Guangzhou Geneseed Biotech Co., Ltd.). All other reagents were of analytical reagent grade and were purchased from Sigma-Aldrich.
Methods
Circularization of ssDNA by T4 Dnl
Based on our previous reports (An et al. 2017; Cui et al. 2018; Sui et al. 2019, 2021), circular ssDNAs were prepared by “One-step strategy” (Supplemental Fig. S2) or “Stepwise strategy” as follows.
One-step preparation
For preparing circular ssDNAs ∼100 nt long, two short 5′-phosphate fragments were used. For a typical reaction, 13.9 µL ddH2O, 1 µL ssDNA fragment-1 (20 µM), 1 µL ssDNA fragment-2 (20 µM), 0.8 µL splint-1 (50 µM), 0.8 µL splint-2 (50 µM), 2 µL 1× T4 Dnl buffer (diluted from 10× stock solution by ddH2O), and 0.5 µL T4 Dnl (5 U/µL) were added into the reaction tube sequentially. The reaction mixture (20 µL) contains 1 µM ssDNA fragments, 2 µM splints, 0.25 U/µL T4 Dnl, and 0.1× T4 Dnl buffer (1.0 mM MgCl2, 50 µM ATP, 1.0 mM DTT, and 4.0 mM Tris-HCl [pH 7.8 at 25°C]). After incubating at 37°C for 4 h, the solutions were increased to 70°C and kept for 10 min (to inactivate T4 Dnl). The reaction solutions were stored at 4°C or −20°C for further experiments.
Stepwise preparation
For those larger circular ssDNAs (>150 nt), three or more fragments were used (Sui et al. 2021). Take the preparation of SSC278 (a 278-nt single-stranded circular DNA) as an example, for which five fragments (B′, C′, D′, E′, and F′, see sequences in Supplemental Table S1) were used; the protocol is as follows (three steps in total). At first, L-B′C′ and L-D′E′ were prepared (10 µL in total, here, L is the abbreviation of Linear). To prepare L-D′E′, for example, 1.5 µL ddH2O, 2 µL D′ (20 µM), 2 µL E′ (20 µM), 3 µL Sp-E′D′ (20 µM, for forming L-D′E′), 1 µL T4 Dnl buffer (10×) and 0.5 µL T4 Dnl (5 U/µL) were added into a PCR tube, then mixed and incubated under 37°C for 2 h. Second, L-D′E′F′ was prepared using L-D′E′ and F′ (10 µL): 5 µL prepared L-D′E′ was transferred to another tube, then, 1.75 µL ddH2O, 1 µL F′ (20 µM), 1.5 µL Sp-F′E′ (20 µM, for ligating F′ to L-D′E′), 0.5 µL T4 Dnl buffer (10×), and 0.25 µL T4 Dnl (5 U/µL) were added. The mixture was incubated at 37°C for 2 h. Finally, C-B′C′D′E′F′ (the desired circular ssDNA) was prepared (10 µL): 2.5 µL L-B′C′ (prepared in the first step), 5 µL L-D′E′F′ (prepared in the second step), 0.75 µL Sp-D′C′ (20 µM), 0.75 µL Sp-B′F′ (20 µM), 0.25 µL T4 Dnl buffer (10×), 0.25 µL T4 Dnl (5 U/µL), and 0.6 µL ddH2O were added into a tube and incubated at 37°C for 4 h. Final concentrations: [L-B′C′] = [L-D′E′F′] = 1 µM, [Sp-D′C′] = [Sp-B′F′] = 1.5 µM, [T4 Dnl] = 0.25 U/µL, 1× T4 Dnl buffer. After heating at 70°C for 10 min (to inactivate T4 Dnl), 0.5 µL Exo I (20 U/µL) and 0.5 µL corresponding 10× reaction buffer (670 mM glycine-KOH, pH 9.5 at 25°C, 67 mM MgCl2, 10 mM DTT) were added and incubated at 37°C for 2 h (to remove the linear ssDNAs).
Preparation of circular dsDNA template
To prepare the template only containing dsDNA with Lk8 and Lk9, Taq Dnl was used at a ligation temperature of 60°C. At first, circular ssDNA, its corresponding short complementary ssDNA fragments and Taq Dnl buffer were mixed, then annealed as follows: After incubation at 90°C for 3 min, cooled to 60°C at a rate of 0.1°C/sec and kept at 60°C for 10 min; then cooled to 20°C at a rate of 0.1°C/sec. For ligation, after the above solution was incubated at 65°C for at least 1 min, Taq Dnl was added as quickly as possible, then kept at 65°C for 2 h. Conditions: [circular ssDNA] = 0.45 µM, [each short ssDNA fragment] = 1.0 µM, [Taq Dnl] = 4 U/µL, 1× Taq Dnl Buffer (20 mM Tris-HCl [pH 7.6 at 25°C], 25 mM KAc, 10 mM Mg(Ac)2, 10 mM DTT, 1.0 mM NAD, 0.1% Triton X-100). The samples were stored at 4°C before analysis. Control experiments without Ligase (or using T4 Dnl to prepare a template containing dsDNA with Lk8–10) were also assembled. Conditions for ligation by T4 Dnl: 0.25 U/µL T4 Dnl, 1× T4 Dnl buffer, 37°C for 2 h (followed by 70°C for 10 min to inactivate T4 Dnl).
Purification of circular dsDNA
The circular dsDNAs were purified by both Exo I and Exo III. Conditions are as follows (20 µL): 1 µL Exo I (20 U/µL), 0.5 µL Exo III (200 U/µL), 1 µL 10× Exo I buffer (670 mM glycine-KOH, pH 9.5 at 25°C, 67 mM MgCl2, 10 mM DTT), and 1 µL 10× Exo III buffer (660 mM Tris-HCl, pH 8.0 at 30°C, 6.63 mM MgCl2) were added into the circular dsDNA solution (10 µL), then incubated at 37°C for 2 h (unless noted otherwise), followed by inactivation at 70°C for 10 min.
Conventional RCT reaction (without RNase H)
Conventional RCT was carried out at 37°C for 4 h (unless otherwise indicated) under the following conditions: 1× transcription buffer (40 mM Tris-HCl [pH 7.9], 6.0 mM MgCl2, 10 mM DTT, 10 mM NaCl, 2 mM spermidine), 2 mM each NTP ([NTP]total = 8 mM), 50 nM dsDNA template, 2 U/µL T7 RNAP, and 2 U/µL RNase inhibitor. Reactions were terminated at 70°C for 10 min.
Cleavage of the RNA product (tandem repeats) with RNase H after transcription (not aided by Helper)
A typical RNase H cleavage reaction was carried out as follows (total volume of 20 µL). Ten microliters of the above transcription solution was sucked out and added to a new tube. Then 8 µL ddH2O, 1 µL 10× RNase H buffer, and 1 µL RNase H (5 U/µL) were added subsequently. The final concentrations were (containing 50% of transcription solution) 0.25 U/µL RNase H and 0.5× RNase H buffer (37.5 mM KCl, 25 mM Tris-HCl, 1.5 mM MgCl2, 5 mM DTT, pH 8.3 at 25°C). After incubating at 37°C for 2 h, the solutions were increased to 65°C and kept for 10 min (to inactivate RNase H).
Remove the dsDNA template after transcription by DNase I
To 35 µL of transcription solution, 21 µL ddH2O, 3.5 µL 10× DNase I buffer, 7 µL DNase I stock solution (1 U/µL), and 3.5 µL RNase inhibitor were added (70 µL in total). Final concentrations: 2 U/µL RNase Inhibitor, 0.1 U/µL DNase I, and 0.5× DNase I buffer (5 mM Tris-HCl [pH 7.5], 1.25 mM MgCl2, 0.05 mM CaCl2). The sample was incubated at 37°C for 4 h, followed by heat-inactivation at 75°C for 10 min.
Cleavage of the RNA product (tandem repeats) with RNase H after transcription (aided by Helper)
To 10 µL of the above solution for DNase I treatment (20 µL volume), ddH2O, 1 µL 10× RNase H buffer, a different set of Helper and 1 µL RNase H (5 U/µL) were added subsequently. Final conditions: 0.25 U/µL RNase H, 0.5× RNase H buffer, 1 µM each Helper. Reactions were carried out at 37°C for 2 h, followed by heat-inactivation at 65°C for 10 min.
Reaction of pfRCT
RNase H (0.25 U/µL final concentration, unless noted otherwise) was introduced into the above “Conventional RCT reaction (without RNase H)” system. In some cases, Helper (ssDNA fragment) was added with the final concentration of 1 µM, and IPP was also introduced (final concentration of 1 U/mL). RNA products were analyzed by PAGE or agarose electrophoresis.
Digestion of RNA product with ShortCut RNase III
A typical ShortCut RNase III digestion reaction was carried out as follows (total volume of 10 µL). After 5 µL of the above transcription solution was transferred to a new tube, 2.5 µL ddH2O, 0.5 µL 10× ShortCut RNase III buffer, 1 µL ShortCut RNase III (2 U/µL), and 1 µL MnCl2 (10×, 200 mM) were added subsequently. Final concentrations (containing 50% of transcription solution): 0.2 U/µL ShortCut RNase III, 1× MnCl2 (20 mM), and 0.5× ShortCut RNase III buffer (25 mM Tris-HCl, 25 mM NaCl, 0.5 mM DTT [pH 7.5 at 25°C]). After incubating at 37°C for 1 h, loading buffer (containing 50% glycerol, 50 mM EDTA, and bromophenol blue) was added to terminate the reaction.
Hybridization of RNA product and ssDNA fragments
A typical hybridization reaction was carried out as follows (10 µL in total). After 5 µL of transcription solution was transferred to a new tube, ddH2O, 0.5 µL 10× RNase H buffer, and short DNA fragments (the final concentration of which was 1 µM) were added. The solution was denatured and annealed as follows: incubating at 90°C for 10 sec, cooling to 65°C (0.1°C/sec), then keeping for 10 min, followed by cooling to 37°C (0.1°C/sec) and keeping for 5 min.
Digestion of the RNA product with the aid of short DNA (SD)
A typical digestion reaction was carried out as follows (10 µL in total). An amount of 5 µL of pfRCT reaction solution was sucked out and added to a new tube. Then ddH2O, 0.5 µL 10× RNase H buffer, different sets of SD (e.g., SD1 + SD3, see sequences in Supplemental Table S1), and 0.5 µL RNase H (5 U/µL) were added subsequently. Final conditions: 0.25 U/µL RNase H, 0.5× RNase H buffer, 1 µM SDs (each with a final concentration of 1 µM), 37°C for 6 h, followed by heat-inactivation at 65°C for 10 min.
Ligation of ssRNA ends on dsRNA by T4 Rnl1
After 10 µL transcription solution was transferred into another tube, 5 µL ddH2O, 1 µL 10× T4 Rnl1 buffer, 1 µL T4 Rnl1 (10 U/µL), 2 µL ATP (10 mM), and 1 µL RNase Inhibitor (40 U/µL) were added subsequently (20 µL in total). Final conditions (containing 50% of transcription solution): 0.5 U/µL T4 Rnl1, 0.5× T4 Rnl1 buffer (including 50 mM Tris-HCl, 10 mM MgCl2, 1.0 mM DTT, pH 7.5 at 25°C), and 1 mM ATP, 2 U/µL RNase Inhibitor, 37°C for 6 h, followed by inactivation at 70°C for 10 min.
Digestion of dsRNA by RNase R for verifying the circularization of free ends of dsRNA
After 10 µL cyclization solution was sucked out and transferred into another reaction tube, 8 µL ddH2O, 1 µL 10× RNase R buffer and 1 µL RNase R stock solution (20 U/µL) were added (20 µL in total). Final conditions: [RNase R] = 1 U/µL, 0.5× RNase R buffer (10 mM Tris-HCl [pH 8.0], 50 mM KCl, and 50 µM MgCl2), 37°C for 1 h, followed by inactivation at 70°C for 10 min.
Evaluation of product concentration and conversion of NTP
The transcription products were analyzed on PAGE (stained with Ultra GelRed) or agarose (prestained with Gene Green). The fluorescence intensity of each band was quantified by Image Lab software. Gel band intensity versus time was then plotted using Origin 8.0. Gel assays were carried out in triplicate.
Determination of product concentration
Based on the band fluorescence intensity of the Ladder with a known concentration (amount), the concentration of transcription product (desired dsRNA) was determined. Here, we suppose that Gene Green has the same ability for staining dsDNA and dsRNA.
Yield of dsRNA (conversion of NTP)
Taking T-B15 as an example, the maximum concentration of dsRNA that could be produced based on the total NTPs (2 mM of each NTP) in the solution is calculated. The numbers of nucleotides T, A, C, and G in the template of T-B15 are 26, 34, 27, and 22, respectively. If all the 2 mM UTP (or ATP) is consumed, the maximum concentration of produced dsRNA (containing 60 uridines and 60 adenosines) should be 33.3 µM. The yield of dsRNA is determined by the ratio of measured dsRNA concentration to this theoretical value (33.3 µM).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The authors are grateful for the financial support by the National Natural Science Foundation of China (32102064 to R.A.), the Shandong Provincial Natural Science Foundation, China (ZR2019BC096 to R.A.), and the Fundamental Research Funds for Co-construction of Universities in Qingdao (X.L.).
Author contributions: H.C. and Z.G. contributed equally to this research; H.C. and Z.G. designed and conducted the experiments; F.L. processed all the pictures, H.C. wrote the manuscript; H.C., Z.G., L.Y., Y.G., R.A., and X.L. revised the manuscript; X.L. conceived the study.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079670.123.
- Received March 25, 2023.
- Accepted July 13, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








