
Is there a localized role for translational quality control?
- 1National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, USA
- 2National Institute of General Medical Sciences, National Institutes of Health, Bethesda, Maryland 20892, USA
- Corresponding author: nicholas.guydosh{at}nih.gov
Abstract
It is known that mRNAs and the machinery that translates them are not uniformly distributed throughout the cytoplasm. As a result, the expression of some genes is localized to particular parts of the cell and this makes it possible to carry out important activities, such as growth and signaling, in three-dimensional space. However, the functions of localized gene expression are not fully understood, and the underlying mechanisms that enable localized expression have not been determined in many cases. One consideration that could help in addressing these challenges is the role of quality control (QC) mechanisms that monitor translating ribosomes. On a global level, QC pathways are critical for detecting aberrant translation events, such as a ribosome that stalls while translating, and responding by activating stress pathways and resolving problematic ribosomes and mRNAs at the molecular level. However, it is unclear how these pathways, even when uniformly active throughout the cell, affect local translation. Importantly, some QC pathways have themselves been reported to be enriched in the proximity of particular organelles, but the extent of such localized activity remains largely unknown. Here, we describe the major QC pathways and review studies that have begun to explore their roles in localized translation. Given the limited data in this area, we also pose broad questions about the possibilities and limitations for how QC pathways could facilitate localized gene expression in the cell with the goal of offering ideas for future experimentation.
Keywords
INTRODUCTION
An exciting discovery to emerge over the last decade is that of “quality control” or “surveillance” sensors that directly monitor the translating ribosome. These proteins perform key roles in triggering a response to acute problems that arise during translation as well as feedback to regulate gene expression and maintain cellular homeostasis (D'Orazio and Green 2021). In this sense, the phrase “quality control” (QC) encompasses a range of outcomes that may be particularly important during stress, development, or other times when the cell's need for protein synthesis changes. These pathways can sense ribosomes that terminate translation prematurely (nonsense-mediated decay, NMD) or arrest during the elongation cycle (ribosome-associated quality control, RQC; the integrated stress response, ISR; and others), and have been recently reviewed in detail (Filbeck et al. 2022; Karousis and Mühlemann 2022). They have multiple outputs that include activation of transcription, mRNA degradation, the molecular resolution of arrested ribosomes, protein modification (phosphorylation, ubiquitination, and ufmylation), and changes to mRNA translational efficiency (ribosome loading).
These QC pathways are generally assumed to be active throughout the cytoplasm. However, this contrasts with the observation that gene expression is not always spatially uniform. Many mRNAs are localized to specific regions in the cell and the efficiency of an mRNA's translation can depend on this location, as discussed in recent reviews (Holt et al. 2019; Das et al. 2021; Gasparski et al. 2022). Localized gene expression enables proteins to be targeted to specific regions where they are most needed (i.e., the endoplasmic reticulum [ER], mitochondria, or axons). In addition, the localization of protein synthesis may have other roles, such as ensuring translation only takes place in regions where there is an appropriate supply of ribosomes or where signaling molecules may be abundant to regulate translation in response to stimuli.
Given the localized nature of protein synthesis for many mRNAs, it is conceivable that the QC pathways that monitor translation could also be locally tuned. Localized activation of QC pathways could, in turn, alter translation efficiency (TE) or mRNA stability to meet the particular needs of a given cellular region. In addition, the transport of mRNAs bound to transiently arrested ribosomes could inadvertently trigger QC pathways, and local tuning could help avoid this outcome. Even global (nonlocal) QC pathways may have roles in localized gene expression if features of localized transcripts are able to make use of them.
Here, we review recent literature and discuss emerging evidence for how QC pathways are important for localized function. As this field is new and studies of localized QC are limited, we also speculate about potential roles and limitations of QC in known cases of localized translation where QC pathways have not been explicitly considered. The intent here is to raise overlooked and thought-provoking questions that we hope will help guide future research directions and lead to deeper insight into the biological importance of localized gene expression. In the interest of simplicity, unless noted otherwise, genes here are referred to by the name of their human orthologs. However, we emphasize that these pathways have roles in many organisms.
THE LIMITS IMPOSED BY DIFFUSION
The importance of localized translation rests on the underlying physics of cells, particularly their dimensions and the rates at which molecules diffuse within them. As such, it is important to keep these basic principles in mind when considering models for how translation, and corresponding QC pathways, can evoke localized effects. Small molecules, including nutrients and chemicals used for signaling, diffuse rapidly and can very quickly equilibrate within a typical ∼20 µm mammalian cell in a matter of milliseconds. Midsized globular macromolecules, such as proteins, can also quickly equilibrate on a slightly longer timescale of 1–10 sec. For example, it was shown that local activation of the eIF2α kinase PKR leads to a wave of visible stress granule formation (a result caused by phosphorylation of eIF2α) as phosphorylated eIF2α diffused across the cell (Corbet et al. 2022). The equilibration of the process was apparent in well under 1 min, consistent with the expected diffusion rate for a protein like eIF2.
In contrast, very large macromolecules, such as mRNAs and ribosomal subunits, diffuse more slowly with a diffusion coefficient of ∼0.1 µm2/sec (Metelev et al. 2022). In these cases, it would take more than a minute to equilibrate throughout the cell. While the establishment of weak gradients with this rate of diffusion is physically possible (Lipkow and Odde 2008), additional mechanisms can augment them so they are more effective for biological functions. Active transport along the cytoskeleton or other mechanisms, such as chemical modification or association with larger cellular structures (anchoring), can facilitate better localization of macromolecules (Buxbaum et al. 2015). Another option to improve localization while diffusion is taking place is to create compartments that impede diffusion, either via a membrane or within protected granules (Moon et al. 2019; Wilbertz et al. 2019). Given these options, it is possible that the availability of critical macromolecules, such as ribosomes or mRNAs, can vary across different parts of the cell.
In neurons, the size scale is larger. While some human neurons can reach up to ∼1 m in length, many neurons have axons that extend ∼1 mm (Boecker et al. 2020). For transport over even 1 mm, however, it is difficult for diffusion alone to move mRNAs or proteins on a useful timescale as it would take hours or days to reach equilibrium (Fonkeu et al. 2019). The neuron, therefore, is a unique environment that requires extensive active transport to facilitate the movement of many kinds of macromolecules.
QC BACKGROUND: A DIVERSE ARRAY OF SENSORS KEEPS WATCH OVER TRANSLATION IN THE CELL
Before exploring the potential roles of localization in QC, we will first review the major features of known QC pathways and their respective sensors (Table 1). While we cannot include every detail, the goal here is to provide an overview of the known breadth of what these sensors detect and what pathways they trigger after they are activated. In particular, we will discuss NMD as well as RQC, the ISR, ufmylation, and several other pathways linked to ribosome stalling, including those triggered by RNF14/RNF25, eIF5A, cGAS, and NOT5. As noted above, we broadly define these pathways to not only include the initial sensing event, but also downstream effects, such as ribosome rescue, activation of transcription, or mRNA degradation. Historically, many of the QC pathways were discovered as a result of their role in mRNA decay and we, therefore include discussion throughout of RNA surveillance (Wolin and Maquat 2019), including NMD, no-go decay (NGD), nonstop decay (NSD), and codon optimality-mediated mRNA degradation (COMD).
Major sensors of stalled 80S ribosomes and disomes
Nonsense-mediated decay regulates gene expression and ensures mRNA quality
One of the earliest QC pathways to be studied plays a key role in RNA surveillance by detecting premature translation termination events and triggering mRNA decay (Fig. 1A). This NMD pathway is present in mammals, yeast, and many other eukaryotes (Kurosaki et al. 2019). It is activated when a ribosome terminates translation prematurely, typically at a stop codon that is far upstream of the 3′ end of the mRNA or near other features that indicate the termination event is premature, such as the exon junction complex (EJC) that remains on mRNAs after splicing (Le Hir et al. 2001; Singh et al. 2008; Hogg and Goff 2010). Notably, most human genes lack EJCs in their 3′ untranslated regions (3′UTRs). Initially, premature termination events are recognized by the NMD machinery, which includes the proteins UPF1, UPF2, and UPF3 (Leeds et al. 1991; Lim et al. 1992; Lykke-Andersen et al. 2000). Degradation of the mRNA is thought to begin cotranslationally (Hu et al. 2010; Kurosaki et al. 2018) with an endonucleolytic cleavage event that is catalyzed by SMG6 and/or by recruitment of decapping and deadenylation factors, such as SMG5 and SMG7 (Lejeune et al. 2003; Unterholzner and Izaurralde 2004; Glavan et al. 2006; Huntzinger et al. 2008; Eberle et al. 2009). While NMD is useful for eliminating aberrant transcripts when a nonsense mutation is introduced into a coding sequence, it is becoming clear that NMD has a broader role in gene regulation by tuning mRNA abundance and eliminating transcripts with regulatory, rather than coding, roles (Kurosaki et al. 2019).
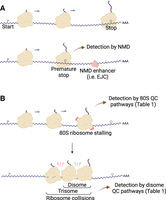
(A) Schematic showing how premature termination triggers the NMD pathway. Premature stop codons can be defined by the large distance separating them from the poly(A) tail and/or their proximity to mRNA binding proteins that enhance NMD, such as EJCs. (B) Schematic demonstrating how ribosome stalling can lead to ribosome collisions, which includes disomes (smallest units) as well as trisomes and higher-order complexes. The extent of ribosome queueing depends on many factors, including TE and the duration of the stalling event. Both stalled 80S ribosomes and disomes can be detected by QC pathways (Table 1). Created with BioRender.com.
Collisions between ribosomes lead to the formation of disomes and signal slow translation
Translating ribosomes can stall due to naturally occurring obstacles found on mRNAs (Tholstrup et al. 2012), defective or damaged transcripts (Yan and Zaher 2019), or the impact of environmental stressors such as nutrient deprivation and oxidative stress (Guydosh and Green 2014; Yan et al. 2019; Rubio et al. 2021; Meydan et al. 2022). Since these events may be a sign of distress or other important biological events, it has been a longstanding question to answer how the cell detects them. In many cases, the unique signature that is detected is the formation of a “disome” (di-ribosome) (Wolin and Walter 1988) complex from the stalled ribosome and an upstream, translating ribosome that collides (bumps) into it (Fig. 1B; Guydosh and Green 2014; Simms et al. 2017; Juszkiewicz et al. 2018; Ikeuchi et al. 2019). If additional upstream ribosomes collide into the disome, higher-order queues (e.g., trisomes) can also form (Matsuo et al. 2020; Meydan and Guydosh 2020). Many recent studies have now established that the interface between the collided ribosomes in the disome complex can be detected by a remarkable number of unique sensors in the cell (Table 1) which, in turn, serve as the trigger of a multitude of downstream signaling events. In this way, the disome is the basic unit of collided ribosomes that is specifically sensed by multiple pathways in the cell. Whether a given stalled 80S ribosome results in a ribosome queue depends on the strength of the stalling event and how well the mRNA is loaded with ribosomes (Hersch et al. 2014). We note that these lists of sensors are not entirely mutually exclusive. In some cases, disome sensors may be somewhat activated by 80S monosomes; sensors of 80S monosomes may be able to detect the individual ribosomes within a disome complex.
The RQC pathway responds to collided ribosomes
One disome-detection system is the RQC pathway, the details of which have been recently reviewed (Filbeck et al. 2022). This pathway is triggered when the E3 ubiquitin ligase, ZNF598 (Hel2 in yeast), ubiquitinates specific ribosomal proteins within a disome complex (Brandman et al. 2012; Juszkiewicz and Hegde 2017; Matsuo et al. 2017; Sundaramoorthy et al. 2017). The ubiquitination event prevents the stalled ribosome from resuming translation and leads to its rescue (removal from the mRNA) by the RQC trigger complex that includes ASCC3 (Slh1 in yeast) (Matsuo et al. 2017; Juszkiewicz et al. 2020b; Matsuo et al. 2020; Hou et al. 2023). The peptidyl-tRNA remains associated with the 60S subunit and is then targeted for degradation, a process involving many proteins, including LISTERIN and NEMF, and includes the addition of carboxy-terminal Ala and Thr residues that serve as a degron signal (CAT-tails). The 60S and 40S ribosomal subunits are used for further translation after removal of the ubiquitination marks (Bengtson and Joazeiro 2010; Shen et al. 2015; Sitron et al. 2017; Garshott et al. 2020; Meyer et al. 2020). These events can also lead to the decay of the mRNA through pathways called NGD and NSD (Frischmeyer et al. 2002; van Hoof et al. 2002; Doma and Parker 2006; Guydosh and Green 2014; Guydosh and Green 2017; D'Orazio et al. 2019; Ikeuchi et al. 2019; Simms et al. 2019; Glover et al. 2020), which requires additional ribosome rescue events carried out by PELO (Dom34 in yeast) and associated GTPases (D'Orazio and Green 2021).
The ISR also responds to collided ribosomes
Ribosome collisions can also serve as a trigger of the ISR. The ISR is typically activated by phosphorylation of eIF2α by the kinases GCN2, PERK, PKR, or HRI that are found in mammalian cells (Pakos-Zebrucka et al. 2016; Costa-Mattioli and Walter 2020) and respond to different stresses. Whether additional kinases carry out this function remains a topic of ongoing investigation (Lu et al. 2021). Among these, GCN2 is the only kinase conserved in budding yeast (Gcn2) and has been traditionally studied under conditions of amino acid starvation (Dever et al. 1992). Gcn2 activity requires two coactivator proteins in yeast: Gcn1 (GCN1 in human) and Gcn20 (ABCF3 in human) (Marton et al. 1993; Vazquez de Aldana et al. 1995). Activation of GCN2 is promoted by the ribosomal P-stalk, consistent with the idea that amino acid starvation leads to an increase in uncharged tRNAs and that the resultant slowdown in elongation by ribosomes then activates GCN2 (Harding et al. 2019; Inglis et al. 2019; Gupta and Hinnebusch 2023). Moreover, GCN2 was reported to be activated when the ribosome was slowed in a way that did not change uncharged tRNA levels, further pointing to the importance of ribosome stalling for activation (Ishimura et al. 2016). GCN2 includes a HisRS domain that binds uncharged tRNA (Wek et al. 1989); however, the outcome of this interaction in the absence of ribosome stalling remains an open question. Several studies of ribosome stalling showed that disomes could trigger the ISR (Wu et al. 2020; Meydan and Guydosh 2021; Yan and Zaher 2021) and structural data clearly revealed that yeast Gcn1 specifically associates with disomes by bridging the gap between the two 80S ribosomes (Pochopien et al. 2021). Phosphorylation of eIF2α leads to global inhibition of translation and activates the transcription of a specific subset of genes necessary for cellular survival under stress. Since translation initiation is inhibited, a natural consequence of ISR activation is thought to be the reduced probability of ribosome collisions due to diminished ribosome loading of mRNAs.
Several factors have been proposed to regulate GCN2, and therefore activation of the ISR, potentially by affecting GCN2's ability to sense disomes. Two proteins that form a complex (Ishikawa et al. 2005) and can repress Gcn2 in yeast (Wout et al. 2009), Rbg2 (RWDD1/DFRP2 in mammals) and Gir2 (DRG2 in mammals), were observed in the yeast disome structure and could therefore prevent productive binding of Gcn2 (Pochopien et al. 2021). In addition, IMPACT (Yih1 in yeast) is another protein that can suppress the activity of GCN2 by competing for GCN1 binding (Kubota et al. 2000). Interestingly, IMPACT can also bind actin monomers in the cell (see further discussion below), which may regulate its interaction with GCN1 (Sattlegger et al. 2004). Like GCN2, DRG2 and IMPACT possess an RWD domain that is thought to mediate their interaction with GCN1 (Silva et al. 2016; Pochopien et al. 2021).
Recent studies have suggested that many other proteins could affect disome surveillance. For example, the complex of yeast proteins Rbg1 (DRG1 in mammals) and Tma46 (DFRP1 in mammals) is thought to prevent ribosome stalling, which could also affect ribosome collisions (Zeng et al. 2021). Rbg1 is homologous to Rbg2 and can bind Gir2, suggesting that it could also have additional regulatory roles. Intriguingly, many proteins have been proposed to interact with GCN1 and could therefore offer further regulatory potential (Castilho et al. 2014).
In cases where ribosome collisions are persistent or long-lived, they are detected by the ZAKα kinase in mammalian cells. ZAKα activates p38 and c-Jun amino-terminal kinase (JNK) stress pathways and enhances ISR signaling (Wu et al. 2020). These regulatory mechanisms lead to apoptosis of cells via p38 and JNK when the ISR is overwhelmed and cannot promote survival, suggesting a tiered recognition of ribosome collisions based on their severity. Some activation of ZAKα may also occur without disome formation, offering the potential of additional levels of regulatory power (Vind et al. 2020b; Snieckute et al. 2022). The transcriptional responses linked to ZAKα have also been considered an independent stress pathway, the ribotoxic stress response (RSR), that may be associated with particular stressors (Iordanov et al. 1997; Vind et al. 2020a). In addition, recent evidence suggests ZAKα activation by disomes can turn on other transcriptional pathways, including those regulated by YAP (Silva et al. 2022).
Additional sensors of stalled ribosomes
Stalled ribosomes were shown to activate the proteins GIGYF2 and 4EHP that inhibit further translation initiation on the mRNA as a way to limit additional ribosome queuing (Hickey et al. 2020). These proteins were shown to be recruited to ribosome collisions by another disome-specific sensor, the protein EDF1 (Mbf1 in yeast) (Juszkiewicz et al. 2020a; Sinha et al. 2020). In this way, ribosome collisions can locally limit new initiation and reduce the odds of additional ribosomes joining the collision complex. GIGYF2 orthologs in yeast (Smy2 and Syh1) are also associated with the decay of collision-inducing mRNAs (Hickey et al. 2020; Veltri et al. 2022). Ribosome collisions increase the probability of frameshifting (Simms et al. 2019) and Mbf1, as well as Gcn1, Gcn20, and Slf1, were shown to prevent collision-induced frameshifting in yeast (Wang et al. 2018; Houston et al. 2022; Jennings et al. 2023).
Another proposed disome sensor, cGAS, is well known for its ability to bind DNA and shuttle between the nucleus and cytoplasm to promote antiviral outcomes. However, a novel role in binding collided ribosomes was recently shown to induce interferon-stimulated gene (ISG) expression to promote the innate immune response (Wang et al. 2018). Curiously, the disome sensor ZNF598 has also been linked to an antiviral response (DiGiuseppe et al. 2018; Sundaramoorthy et al. 2021), but it is unclear if the pathways interact.
In addition, the ubiquitin ligases RNF14 (Itt1 in yeast) and RNF25 bind to collided ribosomes with GCN1 and ubiquitinate (leading to degradation of) the proteins eEF1A (Oltion et al. 2023) or eRF1 (Gurzeler et al. 2023; Oltion et al. 2023). Degradation of these proteins can be induced with a particular natural product or synthetic compounds, but the physiological signals that trigger this pathway remain unknown. Intriguingly, mutation of eRF1 leads to its degradation in a RNF14-dependent manner (Oltion et al. 2023), and Itt1 was known to be involved in readthrough of stop codons (Urakov et al. 2001), suggesting the pathway may be sensitive to slow termination. Interestingly, a recent study where the footprinting of GCN1-bound ribosomes was performed suggested GCN1 may also have a role in sensing ribosome collisions at nonoptimal codons and that the major output of these events is the recruitment of the CCR4–NOT complex to promote mRNA degradation (Muller et al. 2023). This seemed to be particularly important when ribosomes read past stop codons and translated nonoptimal codons in the 3′UTR. These studies show potentially novel roles for GCN1 beyond that established as an activator of the ISR via GCN2. Consistent with this, RNF14 and RNF25 have RWD domains, much like GCN2, DRG2, and IMPACT as noted above, that mediate the interaction with GCN1 (Oltion et al. 2023), suggesting they may compete with GCN2 for binding to GCN1.
Some additional sensors of ribosome stalling have not been shown to be specifically sensitive to the formation of ribosome collisions. For example, CNOT3 (Not5 in yeast) monitors the ribosome E site and responds to slow decoding by promoting mRNA decay (Buschauer et al. 2020). In this way, suboptimal codons that slow the ribosome also tend to reduce the half-life of mRNA in the cell and therefore modulate gene expression, a process referred to as COMD (Bae and Coller 2022; Wu and Bazzini 2023). In contrast, eIF5A also monitors the E site for cases of slow peptidyl transfer (particularly at Pro codons) and promotes continued elongation by augmenting the ribosome's ability to carry out the peptidyl transfer step (Schmidt et al. 2016; Schuller et al. 2017). In another example, the formation of a disome is prevented by the binding of NFX1 (Fap1 in yeast) to the 80S ribosome, and this binding eventually leads to polyubiquitination by RNF10 (Mag2 in yeast) and degradation of the 40S subunit (Sugiyama et al. 2019; Garzia et al. 2021; Li et al. 2022b). In this way, the cell appears to have specific responses to different classes of stalled ribosome. In an additional case of ribosome ubiquitination, the E2 ubiquitin conjugase Rad6 (UBE2A in humans) facilitates ubiquitination of both 40S and 60S subunits (Simoes et al. 2022), and this redox control of translation by the ubiquitin (RTU) pathway plays a role in controlling ribosome stalling and activation of the ISR under oxidative stress (Meydan et al. 2022).
To summarize, the last decade has revealed a rich collection of sensors that are carefully attuned to the translation process (Table 1). As such, premature termination (NMD) or ribosome stalling, particularly when it leads to disome formation (RQC, ISR, and other pathways), serve as important diagnostic signaling events about the current state of the cell.
LOCAL QC
The QC pathways summarized above were generally found by using approaches that are either global in nature or rely on a reporter mRNA that does not specifically localize in the cell and therefore lack the ability to determine whether these QC pathways have a role in localized translation. However, several recent studies have begun to address this gap by examining the roles of QC pathways in local translation (Fig. 2).
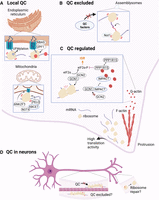
Diagram of a human cell showing examples of how QC can be localized to one organelle in the cell, excluded from particular areas, or locally regulated by the cytoskeleton. Each of the three sections corresponds to a particular subject heading within the manuscript. (A) Local QC. Stalled ribosomes associated with the ER and mitochondria are regulated by dedicated QC pathways. Here, we show the examples of (1) UFMylation of stalled ribosomes and NMD via recruitment of UPF1 by NBAS on the ER, and (2) modulation of the RQC response on the mitochondrial surface by ANKZF1 and NOT4-mediated modification of ABCE1. (B) QC excluded. RNA granules protect mRNAs by shielding them from QC proteins. Here, we include the example of Not1-containing-assemblysomes where proteasome assembly takes place. The granules allow interactions between nascent chains on paused ribosomes to occur without detection by QC factors. (C) QC regulated. A protrusion at the bottom of the cell shows enrichment of ribosomes that support increased translation of mRNAs that are transported to these regions. This phenomenon occurs in different cell types with complex morphology, such as neurons and intestinal cells. Dynamics of actin polymerization are shown as an example of how eIF2α phosphorylation (and ISR activation) could be regulated. Phosphorylation occurs as a result of GCN1-sensed ribosome collisions and more frequent collisions may be expected in this region due to high ribosome loading. Competition between GCN2 and IMPACT for GCN1 would limit this phosphorylation. (D) Examples of QC with localized roles in a neuron. Shown are the examples of QC factors being excluded from sensing stalled ribosomes during transport in axons and potential repair (a type of QC) of damaged ribosomes in the end branches of the axon. Created with BioRender.com.
ER-specific responses to premature translation termination and stalling
Translation at the ER membrane is thought to differ from translation in the cytoplasm in ways that may exclude particular cytoplasmic factors (Reid and Nicchitta 2015). For example, translation of mRNAs usually begins before they are localized to the organelle in order for recognition sequences in the nascent peptide to be detected by signal recognition particle (SRP) or other mechanisms (Ng et al. 1996; Ast and Schuldiner 2013). However, failure of the nascent peptide associated with the translated mRNA to reach the membrane would be detrimental since membrane proteins would be released into the cytoplasm. Once at the membrane, translating ribosomes associate with the translocon in order to thread the nascent peptide into the ER (Voorhees and Hegde 2015; Gemmer et al. 2023). However, this association could obscure parts of the ribosome from being sensed by QC factors or block ribosomes from directly colliding. In addition, ribosome stalling at this stage would be particularly detrimental since it would block a translocon, making it unavailable for further translocation events. The ER membrane, therefore, is a potential hotspot for localized regulation of QC (Fig. 2A).
The NMD pathway includes multiple examples of ER-specific regulation. NMD is known to be regulated by proteins that bind to the mRNA and promote the process by which the UPF proteins detect premature termination events (Kishor et al. 2019). Consistent with this idea, NMD has been reported to rely on ER-specific accessory factors for recruitment of UPF1, particularly the protein NBAS (Fig. 2A; Longman et al. 2020). Similarly, suggestive of ER-specific roles for NMD, a splice variant of UPF1 (UPF1LL) was shown to favor mRNAs that are localized to the ER (Fritz et al. 2022).
There are important consequences when ER-bound ribosomes stall during the elongation cycle since they cotranslationally thread nascent proteins through the ER. If ribosomes stall while associated with the ER translocon, the amino terminus of the nascent protein would be positioned inside the ER lumen, blocking the translocon from processing new protein and increasing stress on the ER. For some types of translation events, it is clear that components of the cytosolic RQC machinery can help resolve these stalled ribosomes, as shown in several studies from yeast. In particular, Dom34 and Hbs1 in yeast (PELO and HBS1L, respectively, in humans) promote ribosome rescue (Izawa et al. 2012). Proteins that act further downstream in stall-detection pathways, such as Ltn1 (LISTERIN in humans) and Vms1 (ANKZF1 in humans), are also involved on the ER by promoting the degradation of the nascent chain via the proteasome (Crowder et al. 2015; Arakawa et al. 2016; Izawa et al. 2017). However, it remains unclear how widespread these roles are and whether the proteins that directly sense disomes, such as ZNF598 or GCN1, contribute to this process on the ER. Interestingly, the disome sensor Hel2 (ZNF598 in humans) was shown to have an important role in detecting ribosomes that tend to arrest after the failure of SRP to recognize its target peptide sequence (Matsuo and Inada 2021). Without detection by Hel2, these mRNAs generate aggregation-bound protein or get mistargeted to the mitochondria (Costa et al. 2018), thus showing an important role for Hel2 in localized translation.
In contrast to the idea of adapting cytosolic QC pathways to function on the ER, recent studies offer evidence of a dedicated QC pathway in mammalian cells that is important for ensuring the quality of secreted proteins (Fig. 2A; Walczak et al. 2019; Wang et al. 2020). This pathway specifically targets ER-bound ribosomes when the UFL1 enzyme modifies uL24 with the ubiquitin-like protein, UFM1. This “UFMylation” of the ribosome occurs when ER-bound ribosomes encounter sequences that trigger ribosome stalling or when cells are treated with anisomycin to globally stall translation (Wang et al. 2020). Additional proteins, such as SAYSD1 (Wang et al. 2023), UFBP1 (Ishimura et al. 2023), and the RQC proteins LISTERIN and NEMF (von der Malsburg et al. 2015; Scavone et al. 2023; Wang et al. 2023), have been proposed to be important for subsequent steps of this pathway. While the involvement of RQC proteins suggests some commonality or overlap with the cytosolic RQC pathway, the observation that nascent chain degradation can make use of the lysosome further points to this pathway being distinct from cytosolic RQC, which relies on the proteasome (Wang et al. 2020). It is conceivable that an ER-specific QC pathway for ribosome stalling is important since the nascent chain may not have a ready opportunity to undergo downstream steps in the canonical RQC pathway due to its location in the ER lumen. In addition, ER-bound ribosomes may be less able to collide and form a stable disome complex (which is required for many cytosolic QC pathways), given the geometry of the ribosome's association with the translocon. This unique geometry could influence how additional steps in translation take place, such as ribosome recycling (Young and Guydosh 2022), the process that removes the ribosome from the mRNA (and presumably the translocon) after translation termination. How exactly UFMylation impacts the detection and resolution of stalled translation on the ER remain important questions for further study.
The ER-resident endonuclease IRE1 has been long known to sense stress from misfolded proteins and trigger the unfolded protein response (UPR). IRE1 facilitates activation of the XBP1 transcription factor, which induces genes to help alleviate folding stress, but also triggers an RNA decay pathway known as regulated IRE1-dependent decay (RIDD) (Hollien and Weissman 2006). RIDD leads to endonucleolytic cleavage within mRNAs encoding many ER-resident proteins. When ribosomes reach the 3′ ends of the cleaved mRNAs, they stall and invoke the NGD pathway (downstream from RQC), as observed in fission yeast (Guydosh et al. 2017). Components of this pathway, including the ribosome rescue proteins Dom34 and Hbs1 (PELO and HBS1L in humans), were shown to be important for fitness under folding stress. Translation on the ER therefore has a particular need for the NGD to enable this regulation. Given the broad roles for these ribosome rescue factors throughout the cytoplasm, it would appear that their availability is widespread, both in the vicinity of the ER and away from it.
Mitochondrial-specific responses to ribosome stalling
Mitochondria are another hub for localized translation, a topic that has been recently reviewed (Wang and Ye 2020), and several QC pathways are known to be active there (Fig. 2A). Cytosolic ribosomes can associate with the mitochondrial surface by interacting with the translocase of the outer membrane (TOM) complex (Gold et al. 2017). These mitochondria-associated ribosomes then locally synthesize mitochondrial proteins and cotranslationally transfer them into mitochondria. Intriguingly, the translation efficiency (TE, a metric of ribosome loading determined by taking the ratio of ribosome footprints to mRNA-seq for a given transcript) of many mRNAs was observed to be enhanced by mitochondrial association (Tsuboi et al. 2020). The authors offered several possible hypotheses to explain this, including the local enrichment of ribosomes or specific proteins to promote translation. An important question for future work to address is whether this tuning of TE changes the odds of chance ribosome collisions and the role of QC pathways on mitochondria.
As in the case with the ER, some proteins in the cytosolic RQC pathway that act after detection of a stalled ribosome were shown to be important on the mitochondria in yeast, including Ltn1 (LISTERIN in humans), Vms1 (ANKZF1 in humans), and Dom34 (PELO in humans), particularly for avoiding the toxic effects of nascent peptides with CAT-tails inside the mitochondria (Izawa et al. 2012, 2017). Interestingly, Vms1 becomes enriched on mitochondria during mitochondrial stress and its loss impacts the growth on glycerol (Nielson et al. 2017; Zurita Rendon et al. 2018). These data suggest that it has an important role locally on mitochondria in resolving ribosomes stalled on TOM complexes.
In another study, it was shown that when a mitochondrion is chemically damaged, ribosomes at the surface stall due to blocked translocation and bind proteins used by QC pathways, including PELO, ABCE1, and CNOT4 (Wu et al. 2018). CNOT4, in particular, was observed to ubiquitinate ABCE1, which is responsible for recycling (removing) ribosomes from mRNAs after stalling or termination (Young and Guydosh 2022). Ubiquitination of ABCE1 (Rli1 in yeast) appeared to give it a signaling role in promoting mitophagy, but this modification could conceivably inhibit its function as a recycling factor and therefore trigger additional ribosome stalling (Wu et al. 2018; Allen et al. 2021). This work suggests an intriguing mechanism that connects mitophagy with cotranslational QC, but the precise mechanisms of this process, particularly the role of the PINK1 kinase (one of the key players in mitophagy) that can localize to mitochondria, remain unknown.
Role of QC in granule formation
QC pathways may also have roles in forming RNA granules. It was recently reported that downstream components activated by the RQC pathway (LISTERIN and NEMF) are required for the partitioning of some mRNAs into stress granules that are induced by arsenite (Moon et al. 2020). While the precise mechanism for this requirement is not entirely clear, the result demonstrates that QC pathways can play a role in directing the localization of mRNAs under stress. As we learn more about translation and its role in different granule structures in the cell (Mateju et al. 2020; Tian et al. 2020; Fernandopulle et al. 2021; Helton and Moon 2022), it will be important to consider the role of QC pathways in their formation and function.
Summary of localized QC
In the above examples, QC pathways play a critical role in regulating the localization or translation of an mRNA on or within particular organelles, such as the ER or mitochondria. In some of the examples, components of the QC pathways are themselves localized with the mRNA via accessory factors or, as in the case of UFMylation, the key enzyme UFL1 is itself localized. In other cases, it appears the localized translation relies on the general availability of QC proteins throughout the cytoplasm though more work is needed to determine whether localized enrichment or activation ever plays a role.
QC EXCLUSION
The previous section highlighted cases where QC pathways are needed for localized function. However, it is conceivable that there may be scenarios where it is advantageous to inhibit a QC pathway in a localized region to prevent unneeded activation. Inhibition would allow for the occurrence of ribosome stalling, or the formation of collisions, that may be functionally required as a condition of mRNA transport or a cotranslational processing step. Similarly, very high levels of ribosome loading may be required for heavy-duty translation and the attendant collisions that would occur with higher probability as a result could inadvertently trigger a QC pathway. We review recent evidence of QC exclusion in this section and then more broadly speculate about how existing models of localized translation may be influenced by the activity of QC pathways (Fig. 2B).
Protection from QC in granules
Recent studies have offered evidence of localized translation in transiently formed granules that appear critical to ensure the proper function of the synthesized proteins (Fig. 2B). For example, an mRNA granule was shown to facilitate cotranslational assembly of the proteasome components Rpt1 and Rpt2 in yeast (PSMC2 and PSMC1, respectively, in human cells) (Panasenko et al. 2019). Formed upon proteotoxic stress, the authors termed these “Not1-containing-assemblysomes” since they were enriched with Not1 protein (CNOT1 in human cells). The authors showed that assembly requires ribosomes pausing at specific regions of the yeast RPT1 and RPT2 mRNAs (also apparent in data from human cells) that would be expected to allow key regions of the nascent peptides to emerge from the ribosomal exit tunnel, presumably without folding. Once the interaction of Rpt1 and Rpt2 is established, translation is again reactivated. While it is not known whether these functionally important stalling events lead to ribosome collisions or recognition by the RQC machinery, the authors suggested these granules might be important for limiting access of QC machinery or reducing the rate of translation initiation as a way to prevent ribosome collisions and unnecessary activation of QC (Collart and Weiss 2020). In a later study, the authors also found that granules enriched in Not4 and Not5 in yeast (CNOT4 and CNOT3, respectively, in humans) excluded eIF5A (Allen et al. 2021). Since Not5 and eIF5A both bind to stalled ribosomes (Table 1), the authors proposed that this separation may play a role in regulating elongation and preventing competition for ribosome binding. The study therefore constitutes another case where phase separation of mRNAs may exclude QC factors to allow for regulation of elongation and functional outcomes.
In yeast, it was shown that many highly expressed genes are localized in granules (Lui et al. 2014). In a follow-up study, which also included evidence for the phenomenon in human cells, these granules were termed “core fermentation” (CoFe) granules because they were enriched in mRNAs encoding glycolytic enzymes that were actively translated (Morales-Polanco et al. 2021). A related study identified “translation factor mRNA granules” in yeast bud tips that were also associated with actively translated transcripts and encoded components of the translational machinery (Pizzinga et al. 2019). Given the evidence that granules can regulate access of QC pathways in the case of proteasome mRNAs (above), it is worth exploring whether these granules also offer protection. Shielding from QC pathways that detect ribosome collisions that tend to form by chance on these heavily translated mRNAs (Meydan and Guydosh 2020) could potentially offer some benefit. Consistent with this hypothesis, only some of these granules were remodeled by amino acid starvation, which is known to induce ribosome collisions and the ISR (Lui et al. 2014).
How do transported mRNAs avoid QC?
While many examples of mRNA localization are known, it is not clear what role, if any, QC pathways play in the localization. For example, mRNAs encoding centrosome components are transported to centrosomes and they are translated during transport in cells from many organisms (Sepulveda et al. 2018; Bergalet et al. 2020; Safieddine et al. 2021). This translation was shown to be essential for proper targeting, suggesting a role for the nascent chains (Safieddine et al. 2021). RNA-binding proteins such as FMRP, which is known to be associated with ribosomes (Chen et al. 2014; Richter and Zhao 2021), may also be important for this process (Ryder et al. 2020). One key question raised by this observation is whether there is any mechanism to limit or slow translation until the mRNAs arrive at their destination to avoid premature release of centrosomal proteins. Since slowing translation runs the risk of triggering QC pathways that detect stalled ribosomes, it will be important to examine the effect (or exclusion) of these pathways in the translation of these localized mRNAs.
This question of how transported mRNAs interact with the QC machinery is particularly relevant in neurons since local translation following transport is critical for growth cone guidance (Yao et al. 2006). One strategy for transporting mRNAs from the soma to their destinations in neurites was reported to involve the repression of translation elongation (Graber et al. 2013; Langille et al. 2019). As in the cases noted above for other cell types, one explanation for how stalled polysomes could evade detection by QC pathways is that the granules in which they are transported are shielded from these factors. Such a model is consistent with observations that neuronal mRNAs remain in a protected or “masked” state until stimulation (Buxbaum et al. 2015). In further support, the absence of QC factors in granule-specific mass spectrometry data sets has been noted (Anadolu et al. 2023). Another possibility is that the ribosomes manage to stall without forming interactions between ribosomes, a strategy that would avoid detection by disome-specific sensors of ribosome collisions, such as GCN1. Consistent with this model, some evidence suggests that FMRP may help “package” stalled ribosomes on mRNAs to prevent the formation of interactions, and therefore collisions, during transport (Anadolu et al. 2023). In addition, evidence that mRNAs can “hitchhike” on organelles, such as lysosomes, raises the possibility that some granules could take advantage of specific environments of associated organelles to regulate activation of QC pathways during transport, particularly in neurons (Liao et al. 2019). A recent report that analyzed the structure and composition of RNA granules derived from rat brain (Kipper et al. 2022) revealed the presence of RNase-resistant disomes in the granules. This observation suggests the disomes could be protected from detection within the granule, particularly as the granules also exhibited evidence of prior lysosome association. Intriguingly, the data also showed the disome sensor EDF1 associated with ribosomes in the granule. Granules may not, therefore, block all QC pathways and EDF1 could potentially have a role in localization.
Beyond the case of transported mRNAs, multiple studies have proposed that the induction of translation can be linked to direct interactions between membrane receptors and the translational machinery in neurons (Koppers et al. 2019). In particular, the DCC receptor, a protein involved in neurodevelopment, was proposed to interact with 80S subunits “poised” to begin translation on start codons (Tcherkezian et al. 2010) and this raises the question of how the arrested 80S resists detection by QC pathways that sense stalled ribosomes. As more studies address the mechanism of extracellular cue-driven initiation of translation in neurons, it will be important to also explicitly consider the role of QC pathways.
Do regions of heavy localized protein synthesis need or exclude QC pathways?
Transcripts encoding ribosomal proteins have been observed to localize in the protrusive fronts of mesenchymal-like migratory cells along with the translational machinery to meet the high demand for translation (Mardakheh et al. 2015; Dermit et al. 2020). Data from pulsed-SILAC mass spectrometry showed that the new ribosomal proteins were eventually transported back to the nucleus, presumably for ribosome biogenesis (Dermit et al. 2020). A question raised by this observation is why does the cell specifically synthesize ribosomal proteins in these protrusions? As ribosome biogenesis takes place in the nucleus, there would appear to be no rationale to localize the production of ribosomal proteins to any particular place in the cytoplasm, especially those distal from the nucleus. In a similar example, the arrival of nutrients during feeding of enterocytes in the intestinal epithelium caused mRNAs encoding ribosomal proteins to localize to the apical part of the cell that senses nutrients and to be translated more heavily (Moor et al. 2017). To meet the demand for translation, the authors noted that the cell also transports extra ribosomes to this region and found other transcripts (even those encoding proteins used primarily elsewhere in the cell) preferentially localized to the apical side, presumably to benefit from the high availability of the translational machinery.
Together, these observations suggest a hypothesis where the cell may use signals (e.g., nutrients or active growth) received in one region of the cell to control protein synthesis, thereby creating an area where mRNAs are heavily translated. A key question for such a model is what role do QC processes play? While it might seem natural to enrich QC factors along with the translational machinery in these regions, it is not clear if this occurs since heavy ribosome loading is expected to increase the chance of ribosome collisions and lead to the activation of QC pathways. More investigation is therefore needed to determine whether proteins from the various QC pathways (Table 1) are generally enriched along with the translational machinery, and if QC inhibitory proteins such as IMPACT (discussed in the next section) play a regulatory role.
QC REGULATION BY ACTIN
A mechanism that has been proposed to broadly modulate translation is based on the interaction between actin and translation factor proteins. Since the polymerization and depolymerization of actin are closely associated with localized movement and growth, actin filament dynamics offer a compelling way to regulate translation, and potentially QC pathways, to adapt to changing conditions in the cell (Fig. 2C).
IMPACT
As noted above, the IMPACT protein can inhibit the ability of the GCN2 kinase to be activated by GCN1, a direct sensor of disomes formed by ribosome collisions (Meydan and Guydosh 2020; Wu et al. 2020; Pochopien et al. 2021; Yan and Zaher 2021). In particular, IMPACT has been proposed to compete with GCN2 for binding to GCN1 and this interaction has been proposed to be modulated by actin monomers (Sattlegger et al. 2004; Silva et al. 2016). Studies in yeast and mammalian cells showed that IMPACT (Yih1 in yeast) preferentially binds monomeric over polymerized actin. Polymerization of actin therefore frees IMPACT to bind GCN1 and could prevent the GCN1–GCN2 interaction that activates the ISR (via phosphorylation of eIF2α) in the presence of disomes. IMPACT therefore represents a link between the dynamic cytoskeleton and QC activation. This link raises the intriguing possibility that disome detection by GCN1 could be limited in the locations where actin is highly polymerized, such as the periphery of cells (Fig. 2C). As a result, these regions may be more tolerant to ribosome collisions and able to increase protein output by loading mRNAs with a level of ribosomes that would typically trigger the ISR due to stochastically formed collisions. Organisms could exploit this mechanism by elevating levels of IMPACT in regions where high levels of protein synthesis may be required, such as the growing bud of a yeast cell or in developing neurons. Consistent with this idea, IMPACT was observed to be enriched in brain tissue and its depletion reduced neurite outgrowth (Pereira et al. 2005; Roffe et al. 2013).
Intriguingly, in many examples of mRNA localization, the activation of translation occurs following transport to particular membrane-proximal regions. For example, mRNAs that depend on the APC protein (Mili et al. 2008) for localization to the cell periphery at either extending or retracting protrusions of migrating mesenchymal cells were found to be repressed at the retracting edge (Moissoglu et al. 2019). In addition, other studies have suggested translation near the growing edge of a cell generally supports heavier translation, particularly on β-actin (Wu et al. 2015; Katz et al. 2016). In a similar example from yeast, the ASH1 mRNA is repressed during transport (Deng et al. 2008). Whether regulation of QC pathways by IMPACT or other factors affects these transport and activation events is an important question for further study.
PPP1R15
Another connection between the ISR and actin cytoskeleton has been suggested by data showing the association of domains in the proteins PPP1R15A and PPP1R15B (also referred to as GADD34 and CREP, respectively), with actin monomers (Fig. 2C; Chambers et al. 2015; Chen et al. 2015). The PPP1R15 proteins are regulatory subunits of a larger complex that includes PP1, an eIF2α phosphatase. The PPP1R15–PP1 complex therefore serves as an antagonist of the ISR. These eIF2α phosphatase complexes are stabilized by interacting with actin monomers, leading to repression of the ISR via dephosphorylation of eIF2α. Strikingly, the reduction of actin monomers near the ER via locally induced polymerization was sufficient to inhibit PPP1R15, which is predominantly found in the ER, and activate the ISR (Chambers et al. 2015). This result therefore showed that local actin dynamics have the power to increase phosphorylation of eIF2α, offering a mechanism by which the ISR pathway could be triggered by a local effect.
Balance between competing pathways
Actin polymerization therefore has two contrasting roles in the regulation of ISR: (i) it decreases levels of P-eIF2α by releasing IMPACT and (ii) it increases levels of P-eIF2α by destabilizing PPP1R15 phosphatase complexes. Future work is therefore needed to dissect how the activity of these two opposing pathways is coordinated. Moreover, the neuronal enrichment of IMPACT and ER enrichment of PPP1R15 may alter this balance as a function of cell type or location within cells. Interestingly, many other components of the protein synthesis machinery are associated with the actin cytoskeleton (Kim and Coulombe 2010). For example, actin polymerization inactivates eEF1A, the translation factor responsible for the delivery of aminoacyl-tRNAs (Mateyak and Kinzy 2010), which could affect the speed of elongation and ribosome collisions. These observations raise the possibility that actin modulates local translation at many stages, with multiple potential connections to QC pathways, and its role therefore offers rich fodder for future study.
QC SPECIFIC TO NEURONS
Some of the most striking examples of localized translation have been reported in neurons where the limits of diffusion over long distances make it possible to establish substantial differences in concentrations of RNAs and proteins between different parts of the cell. As noted above, the essential transport of mRNAs with stalled ribosomes poses the conundrum of how to avoid inadvertent triggering of QC pathways during transport. In contrast, we now consider cases where the activity of these pathways is known to be critical for local functions specifically in neurons (Fig. 2D).
NMD
One example where localized QC appears important is the NMD pathway since its components were shown to be enriched in axons. This observation is consistent with the view that the decay of localized transcripts is important for neuronal function (Colak et al. 2013). Variation in the localization of NMD machinery would limit gene expression in areas where NMD activators are present (or mRNA-bound inhibitors are absent). Consistent with a localized role for NMD, loss of NMD in commissural neurons leads to dysregulated neuronal trajectories. This was traced back to the role of the mouse Robo3.2 gene, an NMD target that is degraded only in axons that cross a particular physiological zone (the floor plate). Similarly, the rat Arc gene, which is locally translated in dendrites and is essential for long-term memory was shown to be subject to NMD (Giorgi et al. 2007). Intriguingly, both Arc and Robo3.2 include introns in their 3′UTRs that make them particularly strong targets of NMD due to the presence of bound EJCs. Many other neuronal transcripts were also shown to share this characteristic (Giorgi et al. 2007). By targeting these genes, NMD may limit their expression to a particular part of the neuron. In the most extreme form of this scenario, translation of the mRNA in an area where it was not needed would lead to immediate decay of the mRNA via NMD and repression of gene expression. Alternatively, NMD may not totally block expression of the gene (via degradation) and instead serve to limit translation to just a few proteins from a transcript before degrading it, a reasonable expectation since NMD was reported to require a few rounds of translation before the mRNA was degraded (Hoek et al. 2019). This is consistent with data showing every ribosome that terminates prematurely has the potential to invoke NMD (Durand and Lykke-Andersen 2013; Rufener and Mühlemann 2013). In this way, NMD would facilitate short bursts of protein expression from mRNAs upon their translational activation at their eventual destination in neurites. Consistent with a functional role for this model, it was shown that loss of an EJC component (eIF4AIII) alters synaptic strength (Giorgi et al. 2007). However, additional work is needed to further understand this role of NMD.
RQC and ISR
Neural-specific roles for the RQC and ISR machinery are suggested by data showing that mutations in RQC and ISR proteins are linked to neurodegeneration phenotypes (Kapur and Ackerman 2018). One potential explanation for this association is that neurons are particularly sensitive to defective translation products that accumulate in the absence of the machinery that rescues stalled ribosomes or processes the nascent peptide chains. For example, the accumulation of nascent peptides that undergo the initial step of being marked with CAT-tails, but not later degradative steps, contributes to proteotoxicity in mammalian cells (Choe et al. 2016; Defenouillere et al. 2016; Yonashiro et al. 2016), including those in the brain (Chu et al. 2009), and is linked to disease-associated phenotypes in Alzheimer's disease models (Rimal et al. 2021).
Alternatively, neuron-specific phenotypes could reflect a sensitivity to an improper level of collisions between ribosomes due to the QC pathways being unable to rescue ribosomes or trigger feedback that limits translation initiation (and ribosome loading). For example, GCN1/GCN2 is important for regulating the rate of initiation via phosphorylation of eIF2α while ZNF598 and PELO could be especially critical for facilitating rescue of stalled ribosomes. As a demonstration of the specific sensitivity of neurons to ribosome stalling, recent data showed that mutations in glycyl-tRNA synthetase that lead to Charcot–Marie–Tooth (CMT) neuropathy (a neuromuscular disease) increase ribosome stalling at glycine codons in mice and cultured human cells, and this leads to hyperactivation of the ISR (Mendonsa et al. 2021; Spaulding et al. 2021; Zuko et al. 2021). Similarly, loss of PELO and HBS1L, proteins involved in ribosome rescue during NGD (downstream from RQC), leads to defects in brain development (Terrey et al. 2021). PELO has been shown to interact with alternative GTPase binding partners, such as GTPBP2 that was important for limiting the phenotype associated with the ribosome stalling events noted above for CMT (Zuko et al. 2021). GTPBP2 also was found to resolve ribosome stalling events in mice triggered in the brain by a mutation in the n-Tr20 tRNA that is specific to the central nervous system (Ishimura et al. 2014). Mutations in a homolog of GTPBP2, GTPBP1, were also shown to evoke similar phenotypes, leading to the activation of GCN2 and the ISR in both cases (Ishimura et al. 2016; Terrey et al. 2020). A key question for future study is how the distribution of QC proteins varies between neurites and soma, as this could offer hints about potential roles of ribosome stalling in different parts of the neuron and in transport (as discussed above).
In a particularly intriguing study that highlights the importance of the ISR pathway in neurons, the authors reported that localized phosphorylation of eIF2α (due to ISR activation) could be observed when neurons were subject to the signaling protein Semaphorin-3A (Sema3A), which is known to direct movement in axonal growth cones (Cagnetta et al. 2019). Moreover, global inhibition or activation of the ISR altered the extent of axonal movement in response to a Sema3A gradient, directly linking the locally activated ISR to this movement of the neuron. Moreover, soma-less neurons were also sensitive to ISR inhibition, further demonstrating the local nature of this phenomenon. The mechanism of how local phosphorylation of eIF2α triggered movement remains unclear, particularly as Sema3A also appeared to activate eIF2B (guanine nucleotide exchange factor for eIF2) which antagonizes some effects of P-eIF2α. Nevertheless, this study definitively shows that a localized output of the ISR can direct neuronal movement and raises the question of whether other local triggers of ISR activation, such as ribosome stalling (and collisions) on synaptic transcripts, play any role in axon guidance.
Many studies have now linked multiple QC pathways to neuronal phenotypes. It is possible, therefore, that crosstalk between the pathways may also be important for understanding their impact on neuronal health. For example, the phenotypes for loss of GCN1 and GCN2 in mice are noticeably more severe for GCN1 (a direct sensor of ribosome collisions), suggesting that GCN2 (the kinase of eIF2α) may not be the only downstream effector that is activated when GCN1 senses ribosome collisions (Yamazaki et al. 2020).
Ribosome repair
As noted above for cancer cells and intestinal cells, neurons have also been shown to exhibit localized production of ribosomal proteins (Moccia et al. 2003). But unlike the localized mRNAs in the other cell types, these mRNAs in axons and dendrites are positioned at such a distance from the nucleus that diffusion could not rapidly bring the synthesized proteins back for the biogenesis of new ribosomes. One explanation for this phenomenon was suggested by evidence that these new ribosomal proteins are incorporated into mature ribosomes at sites that are largely surface exposed (Fusco et al. 2021). This observation, and evidence from other studies (Shigeoka et al. 2019), was used to propose that a local repair process for damaged ribosomes exists in neurons as a kind of QC pathway. Such a model offers a rationale for why these proteins would be synthesized in neurites. Further work is now needed to explain what would trigger and facilitate a repair process and whether damaged rRNA also accumulates and is addressed in any way. Recently, in a key study in yeast, ribosome repair of uL16 and eS26 was shown to take place under conditions of oxidative stress, and mechanistic analysis revealed how two chaperones play a critical role in facilitating the exchange of damaged and newly made protein (Yang et al. 2023). This finding establishes how ribosome repair can occur and lays the groundwork for future studies in other cell types. As these proteins were shown to be exchanged in neurons (Shigeoka et al. 2019; Fusco et al. 2021) and the repair chaperones are conserved in human cells (Yang et al. 2023), it is tempting to speculate that this particular mechanism is also at work in neurons.
Variable ribosome loading
The potential existence of a ribosome repair pathway suggests that damaged ribosomes are particularly problematic in neurons. As such, neurites could be especially dependent on QC pathways since aged ribosomes may be more prone to stalling (Stein et al. 2022) during translation and these pathways could resolve them. One way to further reduce the formation of ribosome collisions is to globally reduce translation initiation, and therefore ribosome loading. Recent data suggest this kind of reduction occurs in neurites since measurements of ribosome loading by sucrose gradient sedimentation revealed heavier loading of mRNAs with ribosomes in the cell body than in the neurite (Biever et al. 2020). Further analysis by using the TE metric showed that the level of ribosome loading was not that specific to particular transcripts (Glock et al. 2021), which is broadly consistent with the overall reduced availability of the translational machinery. The cases where specificity was significant may also be explained by reduced availability of ribosomes (Mills and Green 2017; Gaikwad et al. 2021) or particular mechanisms unique to the different compartments. While this reduction in translation may reduce the tendency for ribosome collisions to form, stalled monosomes would still be expected to trigger some QC sensors.
SUMMARY AND OUTLOOK
While the idea of explicitly considering the role of QC pathways in localized gene expression is relatively new, evidence of their influence is already apparent (Fig. 2). It is clear that NMD and RQC have specific roles in regulating local translation on the ER and mitochondria while a dedicated QC pathway on the ER characterized by UFMylation also serves important functions. Recent studies also suggest that there may be occasions to shield mRNAs from QC pathways, particularly in the case of granules where specific cotranslational assembly takes place. In the future, we look forward to finding out whether mRNAs where ribosomes are stalled, including those undergoing transport or poised to receive an extracellular cue, are similarly shielded in any way. We also look forward to studies that will address the question of whether QC pathways are modulated in regions where heavy translation takes place, particularly near the leading edge of growing cells or in nutrient-sensing cells. An attractive mechanism for how QC pathways may be regulated is rooted in the observation that proteins (i.e., IMPACT) involved in QC pathways are known to be linked to the dynamic cytoskeleton. Finally, we also considered the special case of neurons, where the long distances may allow for unique roles for QC pathways, such as NMD, ISR, and RQC in neuronal development and health. We are eager to learn more about the status of QC pathways in axons, particularly with regard to mRNA transport, translational efficiency, and the synthesis of ribosomal proteins that could represent a new type of QC (repair) pathway.
As we look into the future, we are also excited about the promise of new technology to help address many of these outstanding questions. Just within the last decade, single-molecule imaging systems have offered a new method for directly monitoring the translation of single mRNAs across the cytoplasm in real time (Katz et al. 2016; Morisaki et al. 2016; Wang et al. 2016; Wu et al. 2016). These approaches applied to model systems, such as neurons and migrating cells, offer the potential to directly reveal how translation changes during cellular dynamics and over the course of development. More recently, an exciting new technology offers the possibility of imaging the translational status of thousands of mRNAs in single fixed cells and tissues by using an in situ hybridization and sequencing approach (Zeng et al. 2023). This approach also promises to reveal changes in the distribution of ribosomes along mRNAs, and perhaps even queueing, in three-dimensional space. New photo-switchable inhibitors of translation offer additional promise for improved precision (Ko et al. 2022). The use of elongation inhibitors (Aviner 2020) is an important tool for imaging when applied with an awareness of the limits on what such techniques can accomplish (Enam et al. 2020; Hobson et al. 2020). In addition, new low-input methods of ribosome footprinting are promising for bringing the power of the ribosome profiling approach to low-abundance tissues that are challenging to work with, such as neurons (Hornstein et al. 2016; Clamer et al. 2018; Li et al. 2022a; Ferguson et al. 2023; Meindl et al. 2023), and even single cells (VanInsberghe et al. 2021; Ozadam et al. 2023). Other approaches (Gonzalez et al. 2014; Clamer et al. 2021) for isolating active ribosomes or ribosomes from particular tissues will also be important for accomplishing these goals (Silva et al. 2022). In particular, these approaches offer the possibility of performing disome footprinting, an experiment that is important for addressing questions related to the frequency of ribosome collisions that may trigger QC pathways in localized regions.
We anticipate that a deeper understanding of the known pathways (and their crosstalk) will also offer new insights on localized gene expression. For example, the full role of ubiquitination (and deubiquitination) of the ribosome (Dougherty et al. 2020) and additional roles for regulating translation under stress continue to emerge (Meydan et al. 2022). In addition, it is clear that QC pathways may have key roles in detecting aberrant initiation events that involve the scanning 40S ribosomal subunit (Garshott et al. 2021; Garzia et al. 2021). While the focus of existing QC pathways is on termination (Fig. 1A) or ribosome stalling (Fig. 1B), it will be important to fully consider what mechanisms exist to monitor the early (initiation) and late (recycling) steps of the translation cycle. We are only beginning to understand how QC pathways are involved in localized translation, and we eagerly anticipate future advances to uncover new layers of regulation and offer new insight on the etiology of the many diseases that are linked to these pathways.
ACKNOWLEDGMENTS
We thank Voula Mili for helpful discussions and comments on the manuscript. We thank Bobby Hogg for helpful feedback during the writing of this manuscript. We also thank Patrick Smith for helpful suggestions. This work was funded by the Intramural Research Program of the National Institutes of Health (NIH), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (DK075132 to N.R.G.), and the Postdoctoral Research Associate Training Program (PRAT) at the National Institute of General Medical Sciences (NIGMS) (1FI2GM137845 to S.M.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079683.123.
-
Freely available online through the RNA Open Access option.
This is a work of the US Government.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








