
The limits of human microRNA annotation have been met
- 1The Arctic University Museum of Norway, UiT-The Arctic University of Norway, 9006 Tromsø, Norway
- 2Department of Biosciences and Nutrition, Karolinska Institute, 14183 Huddinge, Sweden
- 3Science for Life Laboratory, Department of Microbiology, Tumor and Cell Biology, Karolinska Institute, 17165 Solna, Sweden
- 4Science for Life Laboratory, Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, 10691 Stockholm, Sweden
- 5Department of Genetics, Faculty of Sciences, MNAT Excellence Unit, University of Granada, 18071 Granada, Spain
- 6Biotechnology Institute, CIBM, 18100 Armilla (Granada), Spain
- 7Biohealth Research Institute (ibs. GRANADA), University Hospitals of Granada, University of Granada, 18014 Granada, Spain
- Corresponding author: Bastian.Fromm{at}uit.no
Abstract
Over the last few years, the number of microRNAs in the human genome has become a controversially debated issue. Several publications reported thousands of putative novel microRNAs not included in the curated microRNA gene database MirGeneDB and the repository miRBase. Recently, by using sequencing of ∼300 human tissues and cell lines, the human RNA atlas, an expanded inventory of human RNA annotations, was published, reporting thousands of putative microRNAs. We, the developers of established microRNA prediction tools and hosts of MirGeneDB, raise concerns about the frequently applied prediction and functional validation strategies, briefly discussing the drawbacks of false positive detections. By means of quantifying well-established biogenesis-derived features, we show that the reported novel microRNAs essentially represent false-positives and argue that the human microRNA complement, at about 550 microRNA genes, is already near complete. Output of available tools must be curated as false predictions will misguide scientists looking for biomarkers or therapeutic targets.
Keywords
Since the discovery of the first human microRNAs in 2000 (Pasquinelli et al. 2000) and 2001 (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001), respectively, microRNAs have taken a center stage in human biology, medical research and, in particular, as biomarkers (Wang et al. 2016). Because the microRNA field substantially expanded in the last years, reaching a record ∼16,000 annual publications in 2021 (Kilikevicius et al. 2022), thousands of novel microRNA candidates have continuously been deposited in the microRNA candidate repository miRBase, expanding near proportionally to the number of publications for more than a decade (Fig. 1A; Supplemental File 1). Driven mainly by the advent of next-generation sequencing (NGS) and the development of bioinformatics prediction tools with flexible stringency thresholds (Friedländer et al. 2008; Hackenberg et al. 2009), this expansion in human microRNAs created an imbalance of microRNA complements between closely related species (e.g., human and macaque) that appeared improbable to many researchers given the high conservation and shared biological functions of microRNAs in animals (Fromm et al. 2020).
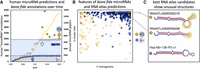
(A) microRNA predictions in miRBase (white), MirGeneDB (blue), and in selected papers (yellow) over time in comparison to the number of annual papers (yellow line) (see Supplemental File 1). A clear near correlation of the number of published papers and annotated microRNAs is detected until 2014 (white arrow, R2 = 0.876), when miRBase introduced high-confidence criteria. The human microRNA complement annotated in miRBase was since then stable at ∼1900 microRNA candidates (light blue area). Note that the curated human microRNA complement (∼500 bona fide microRNAs from MirGeneDB [gray area]) is recovered at 95.2% (480 genes) already by 2010 in miRBase (blue arrow). Several studies predicted substantial numbers of putative novel microRNAs that differ significantly from the putative microRNAs in miRBase and the bona fide microRNAs of MirGeneDB, respectively (yellow dots, see Supplemental File 1; not shown, miRCarta [Backes et al. 2018] with ∼12,000 predictions). Yellow arrow depicts the corresponding numbers for “validated” microRNAs in the RNA atlas from Lorenzi. (B) Comparison of human bona fide microRNA genes as annotated in MirGeneDB (blue) and the novel “validated” microRNA candidates from the RNA atlas (yellow) by mapping of all RNA atlas reads to all precursors (see Supplemental File 2 for summary, Supplemental File 3 for detailed results). Three important annotation criteria based on microRNA biogenesis features were tested: “5′ homogeneity” (x-axis, bona fide microRNAs typically show values beyond 90%), “in cluster ratio” (y-axis, bona fide microRNAs typically show values beyond 90%), and the detection of two precursor arm products that show characteristic RNases derived 2 nt offsets (empty circles: one arm detected, filled circles: two arms detected). Note the few empty circles for MirGeneDB entries that represent either deeply conserved (Hsa-Mir-1307, eutherian), generally lowly expressed (Hsa-Mir-5579), or extremely tissue-specific entries (Hsa-Mir-217, pancreas specific and also deeply conserved) that are detected with both arms in the MirGeneDB data. Pink RNA atlas predictions represent fragments of a previously annotated Y-RNA fragment (R4). (C) The two best RNA atlas candidates as measured by the 3′ feature comparison show strong secondary loop structures (RNAATLASMIR06278), or unusually short loop sizes (RNAATLASMIR06498) in comparison to a representative example from MirGeneDB (Hsa-Mir-126-P2-v1) (red dots mark mature microRNA nucleotides and blue dots star microRNA nucleotides; unusual loop regions are highlighted in yellow).
While it is difficult to define annotation criteria for many RNA classes such as lincRNAs or piRNAs, structure and sequence features of microRNAs are distinct and have been used to develop a system for their annotation already in 2003 (Ambros et al. 2003). However, because these clear and mechanistically well understood features are not all easily implementable in computational prediction (e.g., evolutionary conservation), there is a risk that the number of false positive annotations increases as the community analyzes more data.
With the introduction of stricter criteria for the inclusion of new microRNAs in 2014, miRBase stopped the microRNA expansion for human at a large ∼1900 putative microRNAs (Fig. 1A, white arrow), but kept more than a thousand low-confidence annotations among them (total 295 high and 1586 low confidence) (Kozomara and Griffiths-Jones 2014). Therefore, in 2015, some of us manually curated and reannotated the human microRNA complement by establishing and applying refined criteria for annotation of microRNAs with NGS data (Fromm et al. 2015). Confirming previous experimental work in mouse microRNAs (Chiang et al. 2010), we discovered that two-thirds of human entries were false-positive entries and obtained ∼500 bona fide microRNA genes now hosted in the manually curated microRNA gene database MirGeneDB (Fromm et al. 2015, 2022). When checking previous releases of miRBase, 99% of all bona fide human microRNA genes were already contained in miRBase a decade ago (Fig. 1A), and, with few exceptions, all human microRNAs conserved at least to fish were indeed already found 15 yr ago (Supplemental File 1, “fish human microRNAs in miRBase”; Bartel 2018). Most importantly, despite a range of publications claiming substantial numbers of novel microRNAs in human (Fig. 1A; Jha et al. 2015; Londin et al. 2015; Alles et al. 2019), miRBase and MirGeneDB have only added a few microRNAs to their human complements in the last ∼8 yr, respectively. This held true with the recent release of the telomere-to-telomere assembly of the human genome, where no new microRNAs were found in the previously unassembled regions (Patil et al. 2021).
The idea of creating and updating compendia for gene-products, including noncoding genes such as microRNAs, based on the most recent technological advances is a great service for the scientific community when shared as an online community resource such as the RNAatlas project (Lorenzi et al. 2021). In their “resource paper” Lorenzi et al. used RNA sequencing of small and poly(A) RNA, as well as total RNA, from ∼300 human tissues and cell lines, including cancer cells, to describe at a “more comprehensive atlas of the human transcriptome” including many coding and noncoding RNAs, such as microRNAs. For the prediction of novel microRNAs they used our software miRDeep2 (Friedländer et al. 2012). However, this was done in default settings and without applying the recommended score cut-offs. miRDeep2 output includes numerous statistics, and the score cut-off should be chosen so that the signal-to-noise ratio is above 10:1. Not applying this threshold and not curating the output yielded a staggering number of 3567 novel microRNA predictions (Lorenzi et al. 2021). These were further filtered not by applying well-established annotation criteria (see above, Fromm et al. 2015), but by using “ncRNA target inference algorithms” to identify negative correlations with mRNA to pre-mRNA ratios, obtaining 111 putatively novel microRNAs with inferred targets. While profiling of naïve transcription as compared to overall mRNA abundances has previously been used to distinguish transcriptional and post-transcriptional effects (Tarbier et al. 2020), and anti-correlation of microRNA and overall target expression has been shown to be detectable that is, in single cells (Nielsen and Pedersen 2021), these approaches have their own short-comings and cannot reliably confirm individual microRNA-target interactions. The presence of a few individual pairs showing negative coexpression between microRNA candidate levels and either mRNA levels or proxies for post-transcriptional regulation is to be expected by chance and should not be considered validations for direct microRNA action on its targets and even less as a proof for microRNA-functionality in general. Other approaches such as physiological alterations of microRNA levels, or genetic editing of microRNA target-sites are more likely to capture direct effects (see Huberdeau and Simard 2019; Kilikevicius et al. 2022); however, it is important to note that in either case a functional effect neither validates, nor does its absence disprove microRNA identity.
Because even these 111 filtered novel human microRNAs, as proposed by Lorenzi et al. seemed surprising to us, we systematically tested them for three well-established bona fide microRNA annotation criteria: (A) The “in cluster ratio” which we defined as the proportion of small RNA sequencing reads mapping to the mature microRNA relative to all other reads mapping to a precursor candidate + 10 nt flanking nucleotides (0%–100%), (B) “5′ homogeneity” which is defined as the fraction of mature reads starting at the same 5′ position (0%–100%), and (C) whether reads from both arms of the precursor are detected that show characteristic 2 nt overhang (for review, see Ambros et al. 2003; Fromm et al. 2015; Bartel 2018 on features). To have a comparable and quantitative baseline for these analyses, we took advantage of the manually curated human microRNA complement in MirGeneDB (510 genes excluding variant and noncanonical annotations) and profiled these bona fide microRNAs in all 298 RNA atlas samples, as well.
The results are summarized in Figure 1B (Supplemental Files 2, 3). Briefly, there is an obvious difference between bona fide annotations (MirGeneDB, blue dots) and the RNA atlas predictions. Only two RNA atlas candidates fulfil all three criteria for microRNA annotation (RNAATLASMIR06498 and RNAATLASMIR06278), but all other candidates show either only the expression of one arm, or below average values for 5′ homogeneity and in cluster ratio. However, the two candidates show extremely low expression values (below 5 reads/sample, not shown)—far below what is widely accepted as a biological relevant level (Witwer and Halushka 2016)—and aberrant secondary structures (Fig. 1C): with 51 nt in length, RNAATLASMIR06498 is shorter than any known canonical human microRNA (smallest known are 52 nt in length Mir-374-P1/P2) and with a hitherto not reported short loop of 7 nt. RNAATLASMIR06278 shows exceptional secondary structures in the loop, deviating from the majority of canonical hairpins and warranting further validations (such as the behavior in Drosha- or Dicer-knockdown data [Kim et al. 2016]). In terms of novelty of these rather unlikely microRNA candidates, Lorenzi identified RNAATLASMIR06278 as being annotated in miRCarta, the repository of possible small noncoding microRNA candidates (Backes et al. 2018) (ID hsa-2734-4932.1), but missed that also RNAATLASMIR06498 is found there (ID hsa-5546.1), and thus neither should be seen as novel microRNA candidates.
At first glance it might seem advantageous to include more annotations when looking for biomarkers or therapeutic targets, regardless whether or not they are riddled with false-positives. However, poorly curated microRNA reference and unclear criteria for microRNA annotation can cause substantial downstream issues. For instance, when using a non bona fide microRNA in a model organism as template for the search for partially homologous sequences with supposed biological functions in human (Blanco-Domínguez et al. 2021); when conducting sequence motif searches on a subset of supposed microRNAs that include other RNA fragments (Garcia-Martin et al. 2022); or when interpreting the functions of a snoRNA in the light of microRNA biology (Chinnappa et al. 2022). Further, because small RNA bulk sequencing provides only relative abundance and therefore requires data normalization of expression values, interpretations will be affected by false positives, especially when they are highly expressed (Hamaguchi et al. 2021). Among the RNA atlas candidates are fragments of HY4, a known Y-RNA (Fig. 1B, pink), which show a fivefold higher mean expression than any other candidate. Y-RNAs do not enter the microRNA pathway and therefore can be considered clear false positives (Nicolas et al. 2012). Application of expensive and time-consuming techniques downstream will be in vain when assuming these fragments behave like microRNAs and, thus, the use of a microRNA reference essentially free of false positives is highly recommended for profiling and mechanistic experiments.
In summary, we show that Lorenzi et al., like many other studies before, report large numbers of putatively novel microRNAs which do not represent bona fide human microRNAs, and hence do not advance the field. Prediction tools were developed many years ago when only few data were available and sensitivity for microRNA candidates, but not specificity to bona fide genes was their focus. Because the total number of false-positives increases with the amount of input data, three conclusions can be drawn: First, the output of prediction tools such as miRDeep2 and sRNAbench must not directly be interpreted as novel microRNAs, but instead require stringent interpretation in terms of score cut-off and also manual curation with considerations of conservation, microRNA biogenesis and structural features. Second, for the future, prediction tools need to be adapted to large multitissue input data sets to reduce the number of false positives, thus lowering the downstream manual curation effort; and third, not the repeated resequencing of similar organs, tissues and cell-types, including cancer samples with aberrant expression profiles, but the sequencing of rarely analyzed biological samples or developmental time points might be the source of few additional, hitherto undiscovered microRNAs in human.
We therefore strongly advocate for the careful use of microRNA prediction software for de novo discovery of microRNAs in any organism and reinforce our claim that the human microRNA complement, with ∼550 microRNA genes, is quasi complete.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The authors thank Marc Halushka and Kevin J. Peterson for discussions. B.F. is supported by the Tromsø forskningsstiftelse (TFS) (20_SG_BF “MIRevolution”). M.H. is supported by the Spanish Government (AGL2017-88702-C2-2-R); University of Granada (A-BIO-481-UGR18, FEDER 18).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079098.122.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








