
Artificial mirtron-mediated gene knockdown: Functional DMPK silencing in mammalian cells
- 1Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford OX1 3QX, United Kingdom
- 2Molecular Engineering Laboratory, Science and Engineering Institutes, Singapore 138668
- 3MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, United Kingdom
-
↵4 These authors contributed equally to this work.
Abstract
Mirtrons are introns that form pre-miRNA hairpins after splicing to produce RNA interference (RNAi) effectors distinct from Drosha-dependent intronic miRNAs. Here we present a design algorithm for artificial mirtrons and demonstrate, for the first time, efficient gene knockdown of myotonic dystrophy protein kinase (DMPK) target sequences in Renilla luciferase 3′ UTR and subsequently pathogenic DMPK mRNA, causative of Type I myotonic dystrophy, using artificial mirtrons cloned as eGFP introns. Deep sequencing of artificial mirtrons suggests that functional mature transcripts corresponding to the designed sequence were produced in high abundance. They were further shown to be splicing-dependent, Drosha-independent, and partially dependent on exportin-5, resulting in the precise generation of pre-miRNAs. In a murine myoblast line containing a pathogenic copy of human DMPK with more than 500 CUG repeats, the DMPK artificial mirtron corrected DM1-associated splicing abnormalities of the Serca-1 mRNA, demonstrating the therapeutic potential of mirtron-mediated RNAi. Thus, further development and exploitation of the unique properties of mirtrons will benefit future research and therapeutic RNAi applications as an alternative to conventional RNAi strategies.
Keywords
INTRODUCTION
RNA interference (RNAi) is the cellular process by which short double-stranded RNAs (dsRNAs) mediate translational repression and mRNA degradation in a sequence-specific manner. This pathway has been exploited for therapeutic application with exogenous mimics such as small interfering RNAs (siRNAs) (Elbashir et al. 2001), short-hairpin RNAs (shRNAs) (McManus et al. 2002; Yu et al. 2002), and artificial micro-(ami)RNAs (Zeng et al. 2002). Each approach presents limitations that restrict widespread application, hence novel technologies should be explored (Sibley et al. 2010). Mirtrons, first characterized in flies and nematodes (Okamura et al. 2007; Ruby et al. 2007) and subsequently found in mammals (Berezikov et al. 2007), are natural pre-miRNA intronic hairpins produced through splicing of coding mRNAs, unlike intronic pre-miRNAs, which are produced by Drosha cleavage (Morlando et al. 2008) and have characteristic 3′ overhangs (Fig. 1A). We have previously shown that putative human and murine mirtrons ectopically expressed in HEK and neuronal cells can induce functional knockdown of fully and partially complementary targets (Sibley et al. 2012). This begs the question of whether artificial mirtrons can be used for research or therapeutic RNAi and if they offer any unique differences from current RNAi strategies.
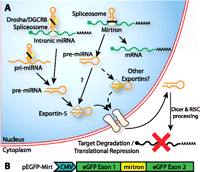
Mirtron biogenesis pathway. (A) Intronic miRNAs are generated with a Drosha-dependent cleavage, while mirtrons are generated by splicing. Because splicing, unlike Drosha cleavage, does not produce defined 3′ overhangs, mirtron export may be mediated by non-exportin-5 pathways. (B) peGFP-Mirt was constructed by inserting cloning sites into a putative exon splice junction in eGFP for the introduction of mirtrons.
Because mirtrons are introns within coding genes, multiple RNAi effectors can be codelivered with a transgene for gene therapy under tissue-specific Pol II promoter control. Exportin-5 can also be oversaturated by shRNAs, negatively affecting endogenous miRNA function (Boudreau et al. 2009; Pan et al. 2011), and even cause severe toxicity in mice (Grimm et al. 2006). Mammalian mirtrons, unlike invertebrate mirtrons or Drosha-processed miRNAs, generally have blunt ends with mismatched last nucleotides (Berezikov et al. 2007) and hence are not predicted to be strong exportin-5 substrates, which have 3′ overhangs (Gwizdek et al. 2003; Okada et al. 2009). Indeed, we have previously shown that miR-877 and miR-1224 have variable dependence on exportin-5 (Sibley et al. 2012). An exportin-5-independent pathway for nuclear export may be able to bypass these molecular bottlenecks. In addition, splicing, being tightly defined by regulatory sequences, yields consistent nucleotides at the ends of the hairpin structures in contrast to Drosha processing in canonical miRNA biogenesis, which can produce heterogeneous ends (Wu et al. 2009), which may lead to off-target effects. These unique differences may present certain advantages over conventional RNAi effectors in specific applications.
Type I myotonic dystrophy (DM1) is a disease caused by an autosomal-dominant unstable CUG expansion in the 3′ untranslated (3′ UTR) region of the myotonic dystrophy protein kinase (DMPK) gene (Mahadevan et al. 1992). Two main mechanisms contribute to disease pathogenesis. The CUG expansion results in nuclear retention of DMPK mRNA and reduced DMPK protein production (Davis et al. 1997), which is essential because DMPK knockout mouse models develop myopathy and cardiac abnormalities (Berul et al. 1996; Jansen et al. 1996; Reddy et al. 1996). The other is sequestration of splice modulators, such as MBNL1, by the CUG hairpin, which results in abnormal splicing of multiple transcripts (Mankodi et al. 2002; Lin et al. 2006). Hence, knockdown of the mutant DMPK gene without abrogating functional DMPK protein is desirable. Discriminating CUG repeat lengths with RNAi for allele-specific silencing is nontrivial, although CAG repeat antisense oligonucleotides appear promising as a therapy (Mulders et al. 2009; Wheeler et al. 2009). Due to the difficulties in discriminating different length CUG expansions in a gene-specific manner, we reasoned that DMPK presents an attractive target for mirtron-mediated RNAi, particularly in light of developing such a gene knockdown and replacement therapy for DM1.
Artificial mirtron design requires consideration of both splicing and RNAi requirements. In this study, we report the development of an algorithm that selects for potential mirtron targets fulfilling these requirements within transcripts of interest. We show that mirtrons designed against human DMPK sequences demonstrate the first reported efficacious RNAi with artificial mirtrons. Further evaluation performed on a subset of mirtrons and compared against their corresponding shRNAs suggests that the efficacy of an antisense designed in two different RNAi systems can differ, underpinning the need for further characterization of the structural determinants of mirtron efficiency. RNA-level knockdown of full-length DMPK constructs was also demonstrated for two mirtrons. This gene knockdown is dependent on splicing, independent of Drosha and partially dependent on exportin-5 transport. Lastly, knockdown of human DMPK in DM500 cells, a model of DM1, corrected splicing abnormalities in Serca-1 mRNA associated with CUG sequestration. This study is the first to report the use of artificial mirtrons for RNAi and demonstrates that this novel technology has the potential to be exploited for research and therapeutic applications.
RESULTS AND DISCUSSION
Development of an artificial mirtron design algorithm
Toward designing artificial mirtrons capable of silencing gene targets, we initially incorporated effective shRNA guide strands into the 3′ arm of miR-1226. However, this predictably did not result in efficient splicing or gene knockdown due to the absence of essential splicing regulatory sequences (data not shown). It was therefore clear that both splicing and RNAi considerations needed to be taken into account when designing mirtron-based vectors. Thus, an algorithm for the selection and scoring of artificial mirtron targets within a gene of interest was developed based on essential splicing requirements, namely, the consensus 5′ splice site (5′ss) sequence and the pyrimidine-rich tract in the 3′ arm, and scored according to previously well-established siRNA design rules (Reynolds et al. 2004) and additional features that can improve splicing efficiency such as an unbroken purine tract that will base-pair with the polypyrimidine tract (Table 1). The scoring system varied according to the hairpin arm (5′ or 3′) location of the antisense/guide stand because of the difference in the number of purines and pyrimidines required. Artificial mirtrons using the loop of mmu-miR-1224 were then designed against human DMPK and introduced as introns within eGFP (pEGFP-Mirt) downstream from a CMV promoter (Fig. 1B). Mismatches were introduced to maximize the number of pyrimidines in the 3′ arm and to bias strand selection by the RISC complex (Khvorova et al. 2003). The target sequences and their corresponding artificial mirtrons are shown in Supplemental Figure S1. In general, the strict requirements for a polypyrimidine tract limits the potential target sequences of a 3′ arm-based mirtron, while the ability to introduce mismatches into the hairpin allows a looser definition of the 5′ arm-based mirtron.
Design rules for selection of artificial mirtron target sequences
Artificial mirtrons targeted against DMPK mRNA sequences are capable of target knockdown
In pEGFP-Mirt, the production of an in-frame transcript is dependent on successful splicing, thus eGFP fluorescence correlates with splicing efficiency. A psiCheck2.2 vector, expressing both firefly and Renilla luciferases, was used as a target vector with one luciferase acting as an internal normalization control for the knockdown of the other in HEK cells. Target sequences were cloned in tandem in the 3′ UTR of Renilla luciferase in dual luciferase constructs, with four perfectly matched targets per construct. Out of 12 artificial mirtron constructs, seven demonstrated appreciable splicing in HEK cells, with two approaching levels of efficiency close to that exhibited by a control human NADH-Coenzyme Q reductase intron 6 (91 nt; NAD) (Fig. 2A,B; Supplemental Fig. S2; Sibley et al. 2012). Luciferase reporter knockdown by the DMPK targeting mirtrons is shown in Figure 2C normalized to the ratio when the NAD intron was used, with eight mirtrons demonstrating significant gene knockdown and four reducing expression >50% of NAD levels. The two mirtrons capable of the greatest reduction in luciferase levels, DMPK-Mirt 5 and DMPK-Mirt 13, were then selected and compared against a U6 promoter-driven shRNA with the same guide strand sequence (Fig. 2D–F). Encouragingly, these demonstrated comparable levels of knockdown to the shRNA at equimolar ratios. Interestingly, greater gene knockdown was achieved with DMPK-Mirt 5 than with its corresponding shRNA, while the opposite was true for DMPK-Mirt 13, similar to how shRNA knockdown efficacy does not necessarily translate to siRNA knockdown efficacy and vice versa (Siolas et al. 2005). Further bioinformatic evaluation of natural mirtron sequences may shed light on the optimal properties for effective artificial mirtron design. This also suggests that if short sequences need to be specifically targeted against, it may be advantageous to examine multiple knockdown modalities.
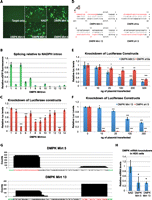
Effective knockdown with an artificial mirtron targeted against DMPK. HEK cells were transfected with 250 ng of psiCheck2.2 targets and 500 ng of mirtron and assayed 2 d later. (A,B) Fluorescence images and quantification of different DMPK mirtrons in a pEGFP-Mirt vector. (C) Knockdown of the luciferase target by DMPK mirtrons. (D) Sequence of DMPK-Mirt 5 and 13 and their corresponding shRNAs. (E,F) Luciferase target knockdown by DMPK-Mirt 5, DMPK5 shRNA after transfection with 250 ng of psiCheck2.2 and increasing amounts of mirtron/shRNA plasmid using the NAD/nonspecific shRNA plasmid to maintain total amount of plasmid transfected (500 ng in total). (G) Deep sequencing was performed with 18- to 25-nt RNA from HEK cells transfected with DMPK-Mirt 5 and 13. Sequences that were aligned to mirtron sequences were collected, 3′ tails were removed, and the sequences were aligned to chart the frequency of individual nucleotides appearing in the small RNA species harvested from the cells. (Red nucleotides) Active guide strand; (green nucleotides) flanking exonic sequences. (H) HEK cells were transfected with 500 ng of phDMPK and 500 ng of mirtron or NAD, and qPCR against hDMPK normalized to 18S RNA was performed on RNA harvested 2 d after. (*) p < 0.05; (**) p < 0.01; (***) p < 0.001 versus NAD/NS shRNA control.
To examine if the processed sequences and strand preference matched mirtron design, high-throughput sequencing was performed on ∼18- to 25-nt RNA from HEK cells transfected with equimolar amounts of DMPK-Mirt 5, 6, 8, 9, 10, and 13. Sequences matched to the mirtrons were then processed, and a histogram of the number of reads containing the specific nucleotides in the mirtron sequence plotted after predicted 3′ tail nucleotides common to miRNAs (Newman et al. 2011) were excluded (Fig. 2G; Supplemental Fig. S3). Agreement between predicted and observed mature mirtrons validates the design methodology. It is also encouraging that strand bias can be very effectively imparted with mismatches on the distal end of the stem–loop. Interestingly, gene knockdown does not correspond to guide strand abundance, which is not surprising given the variable efficacy of siRNA targeting different sequences on the same mRNA; more importantly, the abundance of guide strand does not correspond to eGFP fluorescence, which hints at additional levels of regulation between splicing and the production of mature miRNAs by Dicer. Taken together, this result is the first proof-of-concept demonstration of targeted artificial mirtron-mediated knockdown. In comparison, equivalent DMPK shRNAs designed using well-established principles using the mirtron guide strands result in much higher relative counts of mature short RNAs (Supplemental Fig. S4), as is expected from the much stronger U6 promoter and lack of necessity for processing at the base of the stem–loop. The bias for the 3′ arm of the shRNA in DMPK sh5a was also unexpected but may explain why DMPK Mirt 5 knockdown was stronger than DMPK sh5a.
Following the effective knockdown of luciferase targets, we investigated if artificial mirtrons were capable of knocking down full-length transcripts. Specifically, DMPK-Mirt 5 and DMPK-Mirt 13 were tested for their RNA knockdown effect on ectopic DMPK expression in HEK cells. Importantly, both mirtrons resulted in significant reduction of DMPK mRNA in HEK cells compared with NAD, as measured with qPCR (Fig. 2H). This result suggests the possibility of using these artificial mirtrons against DMPK as a therapeutic strategy for myotonic dystrophy. However, before investigating this further, we sought to define the mirtron origins of these constructs in more detail.
DMPK-Mirt 5 knockdown is dependent on splicing and exportin-5 but not Drosha
To demonstrate that the artificial mirtron activity is dependent on splicing, we mutated the terminal guanosine nucleotide in the 5′ss to an adenosine, which should affect splicing but not silencing efficacy (Fig. 2D). As expected, this abrogated eGFP splicing, as measured by intron-spanning PCR (Fig. 3A) and eGFP fluorescence (Fig. 3B), and inhibited target knockdown (Fig. 3C), demonstrating the dependence on splicing for knockdown. Indeed, this agrees with the deep-sequencing data (Fig. 2G) which also clearly show that the mature guide strand, if within the 5′ arm, has a clearly defined 5′ start site with a near homogeneous 3′ end. Alternatively, if the mature guide strand is within the 3′ arm (DMPK-Mirt 13 only), then there are also clearly defined 5′ and 3′ ends. The uniformity in the 5′ and 3′ ends of the mature strand is probably due to the specificity imparted by pre-mRNA splicing. Because Dicer recognizes its cleavage site with a “molecular ruler” mechanism (MacRae et al. 2007), a well-defined pre-miRNA will likely lead to better Dicer specificity. The homogeneity of the mature guide strand may be particularly advantageous in reducing off-target effects that may arise from imprecise processing of the 5′ end by Drosha using other RNAi strategies (Wu et al. 2009).
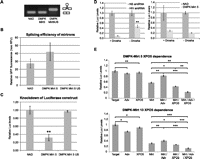
Dependence on Drosha and Exportin-5 for mirtron-mediated knockdown. (A,B) HEK cells were transfected with 250 ng of psiCheck2.2 and 500 ng of mirtron/variant and assayed 2 d later. Splicing efficiency of NAD, DMPK-Mirt5, and DMPK-Mirt5US using RT-PCR with eGFP primers flanking the intron (A) or GFP fluorescence (B). (C) Knockdown of luciferases target by NAD, DMPK-Mirt5, DMPK-Mirt5US, an unspliceable construct. (**) p < 0.01 versus NAD. (D) Knockdown of putative targets in Renilla luciferases 3′ UTR by an shRNA targeting a HIV transcript (positive control) or by DMPK-Mirt5 in HEK cells stably transfected with Lentivirus (MOI = 1) carrying either a nonspecific (+Drosha) or Drosha shRNA (−Drosha), induced for 8 d with 5 μg/mL doxycycline and transfected on Day 6 with mirtron/amiRNA and targets. (**) p < 0.01 between +Drosha versus −Drosha. (E) HEK cells were cotransfected with 250 ng of target with or without 500 ng of mirtron (Mirt), 500 ng of pAdvantage (Adv), and 500 ng of exportin-5 (XPO5). pEGFP-NAD was used to maintain the total plasmid levels (1750 ng per well). Cells were harvested 2 d after and assayed for luciferase activity. (*) p < 0.05; (**) p < 0.01; (***) p < 0.001 versus control.
Next, a lentivirus containing a doxycycline-inducible Drosha shRNA (Aagaard et al. 2007) or a nonspecific shRNA was used to stably transfect HEK cell lines. Dependence on Drosha would result in weaker gene knockdown after Drosha knockdown. However, knockdown by DMPK-Mirt 5 was independent of Drosha knockdown, while a control Drosha-dependent amiRNA against a HIV-1 target based on miR-122 demonstrated decreased knockdown in Drosha-knockdown cell lines (Fig. 3D).
Because pre-mRNA splicing is an efficient and precise process, the probable rate-limiting step in mirtron biogenesis is debranching of intron lariats, mediated by debranching enzyme 1 (DBR1) in humans. However, the kinetics of DBR1 activity are not well known. In the test tube, yeast DBR1 is highly active, taking <1 min to remove all branched lariat derived from the CYH2 mRNA from DBR1-null yeast (Khalid et al. 2005), but expression of a single murine DBR1 cDNA is not sufficient to fully recover the lariat accumulation phenotype of Schizosaccharomyces pombe (Kim et al. 2001), suggesting that the kinetics of mammalian DBR1 are much slower. Another factor that limits debranching is the intrinsic stability of the branched lariat. Certain introns can accumulate as lariats within the nucleus despite functional DBR1 (Qian et al. 1992). A study of Pem homeobox gene introns in HeLa cells suggests that intron half-lives are ∼9–29 min, the shortest being only 9.4 min, and thus intron degradation is rate-limited by the lariat debranching step rather than exonuclease activity (Clement et al. 2001). In support, knockout of yeast DBR1 results in the accumulation of circular lariats lacking their 3′ ends, a result of exonuclease activity (Chapman and Boeke 1991). If DBR1 is a slow enzyme compared with nuclear exonucleases or if artificial mirtrons are unusually stable as lariats, partial digestion of the 3′ end of the lariat could prevent functional hairpin formation or, alternatively, result in variable hairpin formation, which could underlie the difference in mirtron and shRNA efficiency. In support, a recent study has shown that miR-1017, a 3′-tailed mirtron in Drosophila melanogaster, is processed by 3′ exonuclease to form the pre-miRNA (Flynt et al. 2010). Sequencing results of artificial mirtrons may have missed these truncated 3′ sequences because RNA was sized pre-selection, thus further work needs to be undertaken to elucidate the debranching step in mirtron biogenesis and the rules governing the intrinsic stability of introns. This may greatly improve the prediction of optimal mirtron sequences.
Lastly, canonical pre-miRNAs have a characteristic 2-nt overhang on the 3′ arm of the hairpin due to the Drosha cleavage (Lee et al. 2003). This overhang results in the hairpin being the optimal substrate for exportin-5 (Gwizdek et al. 2003; Okada et al. 2009). The general lack of 3′ overhangs in mammalian mirtrons suggests that they may be exportin-5-independent. To investigate if mirtron-mediated knockdown was exportin-5-dependent, we used the pAdvantage plasmid, which expresses VA-1 RNA, an exportin-5 substrate that competes with miRNAs (Lu and Cullen 2004) and overexpression of human exportin-5. Target luciferase constructs were cotransfected with DMPK-Mirt 5 or 13 (Mirt) with pAdvantage (Adv) and/or exporitn-5 (XPO5) (Fig. 3E). The results showed that DMPK-Mirt 5 and 13 knockdowns were adversely affected by VA-1 competition. Since VA1 may also have inhibitory effects on Dicer (Lu and Cullen 2004), we performed rescue assays in the presence of exogenous exportin-5 to see the contribution of this protein to the inhibition seen. These showed that the silencing by DMPK-Mirt 5 and DMPK-Mirt 13 in the presence of VA1 could be improved by the addition of exogenous exportin-5, demonstrating that these mirtrons can use this nuclear export pathway. Furthermore, the combination of exportin-5 and pAdvantage resulted in greater knockdown than with exportin-5 only. This is probably because VA-1 RNA can dramatically improve translation of numerous mRNAs including XPO-5 and, in so doing, improve the efficacy of nuclear export of mirtron. In contrast to previous data with miR-877 (Sibley et al. 2012), a natural mirtron that is completely inhibited by high levels of VA-1 RNA, VA-1 saturation appears to have less pronounced effects on DMPK-Mirt 5– and 13–mediated gene knockdown, suggesting that mirtrons may not exclusively use exportin-5 as the sole nuclear export mechanism. An intriguing nuclear export scenario is that not all mirtronic pre-miRNA hairpins are debranched within the nucleus, and some are exported out of the nucleus as branched lariats. Although the major spliceosome used for artificial mirtron biogenesis is exclusively nuclear (Rogers and Wall 1980), bioinformatic prediction of the human DBR1 protein sequence predicts a cytoplasmic localization (Pierleoni et al. 2006), while lariat debranching activity has also been detected in both cytosolic and nuclear fractions (Arenas and Hurwitz 1987). All spliced introns of the Pem homeobox gene were found to be present in the cytosolic fraction of HeLa cells (Clement et al. 2001). These suggest an export mechanism for branched introns into the cytoplasm from the nucleus. If so, then the export of the branched lariat will probably be independent of exportin-5, and, as a result, mirtron hairpins may be generated in the cytoplasm in addition to being exported out of the nucleus by exportin-5.
DMPK knockdown by DMPK-Mirt 5 corrects splicing abnormalities in DM500 cells
Concurrent knockdown of pathogenic alleles and replacement with RNAi-resistant codon-replaced wild-type genes is a promising therapeutic intervention for diseases caused by dominant mutations of essential genes. The concept has been demonstrated with eGFP, autosomal-dominant retinitis pigmentosa, and HIV using shRNAs (Cashman et al. 2005; Unwalla et al. 2006) and siRNAs (Kim and Rossi 2003; Kiang et al. 2005; O'Reilly et al. 2007). As an alternative to shRNAs, siRNAs, and artificial miRNAs, mirtrons coexpressed within the replacement gene could be driven by the endogenous RNA polymerase II promoter, offering precise spatiotemporal control of knockdown while minimizing the size of the gene cassette such as in the case of DM1. To evaluate the functional applicability of the DMPK mirtrons, we decided to investigate phenotypic effects in a cellular model of DM1.
DM500 cells are murine myoblasts that contain a pathogenic copy of human DMPK with more than 500 CUG repeats (Mulders et al. 2009). Because splicing defects associated with sequestration of MBNL1, including exon 22 exclusion from the Serca-1 mRNA (Lin et al. 2006), are found in differentiated DM500 cells, they provide a robust model for assessing therapeutic correction. DM500 myoblasts were transfected with 1 μg of NAD intron, DMPK-Mirt 5, or DMPK-Mirt 5US in triplicates and differentiated 2 d after transfection. After 7 d of differentiation, the myotubes were imaged and the RNA was harvested. GFP expression was relatively low (Fig. 4A), although significantly greater fluorescence and hence more active mirtron effectors were noted earlier post-transfection. Quantitative PCR analysis of human DMPK mRNA normalized to murine GAPDH shows a significant reduction in pathogenic DMPK (Fig. 4B), suggesting that DMPK-Mirt 5 is capable of knocking down the toxic pathogenic DMPK RNA. Furthermore, RT-PCR of Serca-1 mRNA demonstrated increased inclusion of exon 22 in DM500 cells, suggesting that MBNL-1 sequestration is partially reversed by knockdown of mutant human DMPK mRNA (Fig. 4C). This inclusion of Serca-1 exon 22 after mirtron-mediated pathogenic DMPK knockdown in DM500 cells implies that MBNL-1 has been released from CUG sequestration to mediate normal splicing and provides evidence that mirtron-mediated gene knockdown can be used as DM1 gene therapy.

Functional effects of mirtron-mediated DMPK knockdown in DM500 cells. DM500 cells were transfected with 1 μg of NAD, DMPK Mirt 5, or DMPK Mirt 5US and differentiated 2 d after transfection. After 7 d in differentiation medium, RNA was harvested. (A) Representative GFP fluorescence and merged brightfield and eGFP images of differentiated DM500 before harvest. (B) qPCR of DMPK cDNA normalized to mGAPDH cDNA. (C) Representative RT-PCRs of Serca-1 using primers flanking exon 22. Sequestration of MBNL1 leads to exclusion of exon 22. The graph below shows the relative ratio of cDNA with exon 22 to cDNA without exon 22. (*) p < 0.05 versus NAD control.
CONCLUSION
In this study, we investigated if the endogenous mirtron pathway is exploitable for gene knockdown. A selection algorithm for potential artificial mirtron target sequences was designed, and artificial mirtrons capable of effective RNAi were generated in an eGFP cassette. Selected sequences demonstrated efficacy against native and recombinant target RNA sequences. DMPK-Mirt 5–mediated knockdown was found to be independent of Drosha processing but dependent on splicing and exportin-5, consistent with the biogenesis mechanism of natural mirtrons (Ruby et al. 2007), although exportin-5 may not be the sole nuclear export system used by mirtrons. In terms of therapeutic application, Serca-1 splicing defects were corrected in a cellular model of myotonic dystrophy with DMPK-Mirt 5, suggesting that a therapeutic level of nuclear RNAi was achieved. This suggests that DMPK-Mirt 5 can potentially be used in a gene knockdown and replacement strategy for therapeutic correction of DM1. It will be interesting to see if this construct can successfully be incorporated into a DM1 gene knockdown and replacement therapy by using a construct incorporating multiple DMPK-targeting artificial mirtrons as separate introns within an RNAi-resistant host DMPK transcript in combination with the endogenous DMPK promoter (Storbeck et al. 1998). This is the first demonstration that artificial mirtrons can be exploited for gene knockdown, and this work will now lead to further development of this novel therapeutic tool, in particular for combinatorial gene therapy applications.
MATERIALS AND METHODS
Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich, and all enzymes used were obtained from New England Biolabs.
Plasmids
pEGFP-Mirt and cloning of mirtrons were previously described (Sibley et al. 2012). Briefly, a BbsI-excised sequence was placed between two fragments of eGFP, and introns were introduced in that site with annealed oligonucleotides (Fig. 1B). Luciferase targets were produced by annealing and ligating the relevant targets (Supplemental Table S1) as oligonucleotides into psiCheck2.2 (kind gift from Dr. Marc Weinberg) (Supplemental Fig. S5) downstream from Renilla luciferase between XhoI and NotI sites as previously described by others (Ely et al. 2008). The luciferase targets for DMPK-Mirt1, 4, 5, and 6 were cloned as a concatemer of target sequences, as were targets for DMPK-Mirt 7–10 and DMPK-Mirt 11–14 (Supplemental Table S1).
The relevant shRNAs used for comparison were produced by PCR of the U6 promoter with the reverse complement of the shRNA followed by a reverse U6 primer. The PCR product was then TA-cloned into pCR2.1 (Invitrogen). The final plasmid size is similar to pEGFP-Mirt clones.
Human DMPK amplified from a human DMPK cDNA plasmid (Invitrogen) was cloned into pEGFP-C1 after removing EGFP with NheI and HindIII to produce phDMPK. Human exportin-5 was PCRed from human HEK random nonamer-primed cDNA and cloned into pEGFP-C1 downstream from eGFP using the BspEI and XhoI restriction sites. pAdVantage was obtained from Promega.
Cell culture and transfection
HEK cells were cultured in DMEM Glutamax supplemented with 10% FBS and antibiotics and incubated at 37°C in 5% CO2. DM500 cells (gift from Dr. Wansink) were maintained in proliferation medium (DMEM, 20% FCS, 20 U/mL γ-interferon, 1% antibiotics) in plates coated for 30 min with 0.2% gelatin at 33°C in 10% CO2. For differentiation of DM500 cells, plates were coated with 500 μL of a 1/100 dilution of BD Matrigel (BD #356234), and cells were grown in proliferation medium until confluent, then differentiated with differentiation medium (DMEM, 5% horse serum, 1% antibiotics). Transfection of HEK cells was performed using Lipofectamine 2000 (Invitrogen) in HEK cells and JetPEI (Polyplus Transfections) in DM500 cells as per the manufacturer's instructions in 24-well plates. When the experiment required a control NAD plasmid or shRNA, an amount corresponding to the mirtron plasmid was used. The cells were imaged and lysed 48 h post-transfection.
RT-qPCR and PCR
Reverse transcription was performed with 1 μg of RNA and random nonamers using Expand Reverse Transcriptase (Roche) as per the manufacturer's instructions. qPCR experiments were performed on an ABI7000 thermal cycler in 20-μL reactions, with 30 nM primer and 0.2 μL of each cDNA preparation, using Precision qPCR Mastermix (Primer Design) as per the manufacturer's instructions. Intron-spanning PCR was performed with BioTaq (Bioline) using primers flanking the intron. The primers used for qPCR and PCR are listed in Supplemental Table S1.
High-throughput sequencing
HEK cells were grown in 24-well plates to 80% confluence and transfected with 500 ng of each mirtron or shRNA construction per well. Small RNA libraries were prepared with the Small RNA v1.5 Sample Prep Kit following the manufacturer's instructions (Illumina). Briefly, total RNA was isolated from each transfection by TRIzol extraction and pooled. The RNA was ligated with a 3′-RNA adaptor modified to target small RNAs with 3′-hydroxyl groups, and then with a 5′-RNA adaptor. Reverse transcription followed by PCR was performed to select for adaptor-ligated fragments, and double-stranded DNA libraries were size-selected by PAGE purification (6% TBE PAGE). Libraries were sequenced on a Genome Analyzer IIx for 36 cycles following the manufacturer's protocols. The image analysis and base calling were done using Illumina's GA Pipeline. Adaptors were trimmed with Biopieces remove_adapter script, and remaining sequences were aligned against full-length mirtron hairpins.
Fluorescence quantification and Dual-Luciferase Assay
The luciferase assay was performed with Promega's Dual-Luciferase Reporter Assay System as per the manufacturer's instructions. Briefly, HEK cells were lysed in 100 μL of passive lysis buffer, and background-subtracted eGFP fluorescence was measured in a 96-well format with 20 μL of each sample using Victor3 (PerkinElmer). Twenty-five microliters of Luciferase Buffer II was added to the sample and luminescence was measured. Twenty-five microliters of Stop & Glo Buffer was then added to measure Renilla luciferase activity. The Renilla luciferase signal was then normalized to the firefly luciferase signal for all luciferase experiments.
Microscopy
HEK cells transfected with the mirtron (250 ng) only were imaged 48 h post-transfection with a Leica DMIRB microscope (Leica Microsystems) with a 20× objective to obtain brightfield and GFP fluorescence.
Statistics
All experiments, unless otherwise stated, were performed in triplicate. All error bars used in this report are standard deviations. Statistical significance was determined by a one-tailed Student's t-test assuming equal variance.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Marc Weinberg (University of Witwatersrand) for his valuable discussions and suggestions and Dr. Rick Wansink (Radboud University) for the DM500 cells. The work was funded by Muscular Dystrophy Ireland and the Science and Engineering Council, Agency for Science, Technology and Research (Singapore). Y.S. is funded by the Agency for Science, Technology and Research (Singapore). C.R.S. is funded by the MRC UK.
Author contributions: C.R.S., Y.S., and M.J.A.W. conceptualized the project and wrote the paper. C.R.S. and Y.S. designed, performed, and analyzed all of the experiments.
Footnotes
-
↵5 Corresponding author
E-mail matthew.wood{at}dpag.ox.ac.uk
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.030601.111.
- Received September 27, 2011.
- Accepted May 4, 2012.
- Copyright © 2012 RNA Society








