
In vitro reconstitution of yeast splicing with U4 snRNA reveals multiple roles for the 3′ stem–loop
- Department of Chemistry, University of Northern British Columbia, Prince George, British Columbia, V2N 4Z9 Canada
-
↵1 These authors contributed equally to this work.
Abstract
U4 small nuclear RNA (snRNA) plays a fundamental role in the process of premessenger RNA splicing, yet many questions remain regarding the location, interactions, and roles of its functional domains. To address some of these questions, we developed the first in vitro reconstitution system for yeast U4 small nuclear ribonucleoproteins (snRNPs). We used this system to examine the functional domains of U4 by measuring reconstitution of splicing, U4/U6 base-pairing, and triple-snRNP formation. In contrast to previous work in human extracts and Xenopus oocytes, we found that the 3′ stem–loop of U4 is necessary for efficient base-pairing with U6. In particular, the loop is sensitive to changes in both length and sequence. Intriguingly, a number of mutations that we tested resulted in more stable interactions with U6 than wild-type U4. Nevertheless, each of these mutants was impaired in its ability to support splicing, indicating that these regions of U4 have functions subsequent to base pair formation with U6. Our data suggest that one such function is likely to be in tri-snRNP formation, when U5 joins the U4/U6 di-snRNP. We have identified two regions, the upper stem of the 3′ stem–loop and the central domain, that promote tri-snRNP formation. In addition, the loop of the 3′ stem–loop promotes di-snRNP formation, while the central domain and the 3′-terminal domain appear to antagonize di-snRNP formation.
Keywords
- pre-mRNA splicing
- in vitro reconstitution
- U4 snRNA
- di-snRNP formation
- spliceosome activation
- protein–RNA interaction
- splicing complementation
INTRODUCTION
Splicing, an essential step in eukaryotic gene expression, involves the removal of noncoding introns from precursor messenger RNA (pre-mRNA) and ligation of the flanking protein-coding exons to produce mature mRNA. This process is catalyzed by a large ribonucleoprotein complex, the spliceosome, composed of five small nuclear RNAs (snRNAs)—U1, U2, U4, U5, and U6—and over 100 proteins, some of which associate with a specific snRNA to form the corresponding small nuclear ribonucleoprotein (snRNP). Through a highly ordered and tightly regulated series of interactions, these snRNPs assemble onto the pre-mRNA and, following a number of conformational changes leading to formation of the active spliceosome, catalyze the two chemical steps of splicing.
While each of the five snRNAs plays an essential role in splicing, that of U4 remains the least well understood. U4 and U6 share extensive sequence complementarity (Brow and Guthrie 1988; Guthrie and Patterson 1988), and their interaction in the form of a di-snRNP is required for incorporation of U6 into the active spliceosome (Brow and Guthrie 1988). U4, however, is not directly involved in splicing catalysis and is thought to dissociate from the spliceosome prior to the first chemical reaction (Lamond et al. 1988; Yean and Lin 1991). Furthermore, there has been no direct biochemical identification of the proteins associated with U4 snRNA in the free snRNP, the sequences with which they interact, or the roles they may perform during snRNP and spliceosome assembly.
A powerful tool for examining the function and interactions of the U4 snRNP is in vitro reconstitution. This involves depleting endogenous U4 snRNA from cell extract to abolish splicing activity, followed by complementation with a modified version of U4 to examine the effects of mutations or investigate its interaction partners. Because U4 function is essential, in vitro reconstitution enables analysis of lethal mutations that would not be possible using in vivo methods. In vitro reconstitution assays have been developed for all five mammalian snRNPs (Wersig et al. 1992; Wolff and Bindereif 1992; Ségault et al. 1995; Will et al. 1996), as well as yeast (Saccharomyces cerevisiae) U2, U5, and U6 (Fabrizio et al. 1989; McPheeters et al. 1989; O'Keefe et al. 1996); however, there has been no report of in vitro reconstitution of yeast U1 or U4, though the latter has been attempted (Horowitz and Abelson 1993).
Reconstitution of mammalian U4 has provided valuable insight into the functionally important regions of this molecule. U4 snRNA can be divided into six domains: stem II, the 5′ stem–loop, stem I, the central domain, the 3′ stem–loop (the central stem–loop in mammalian U4), and the 3′ terminal domain containing the Sm binding site (Fig. 1A). By examining deletion mutations within each of these domains, Wersig and Bindereif (1990, 1992) demonstrated that the 5′ portion of U4, including stems I and II and the intervening 5′ stem–loop, is required for U4/U6 di-snRNP formation, subsequent spliceosome assembly, and splicing in vitro, while the 3′ portion of the molecule appeared to be dispensable.
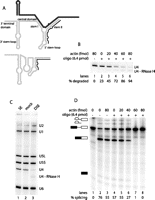
U4 degradation and consequent splicing inhibition require active splicing conditions. (A) Secondary structure of U4 (light gray) base-paired to U6 (dark gray) (Brow and Guthrie 1988) and as a free particle (Myslinski and Branlant 1991), with the targeting oligonucleotide binding site indicated (black line). (B) Extent of U4 degradation increases with actin concentration. Northern blot of U4 in splicing extract in the absence or presence of targeting oligonucleotide and increasing amounts of unlabeled actin pre-mRNA. (C) U4 degradation does not affect other snRNAs. Denaturing Northern blot of untreated splicing extract (SE, lane 1), mock-depleted extract (lane 2), and U4 depleted extract (DSE, lane 3), probed for all five yeast snRNAs as indicated on the side. U5L indicates long U5; U5S, short U5; and U4-RNaseH, U4 cleavage product. (D) Extent of splicing inhibition increases with the concentration of cold actin in the U4 degradation step. Denaturing gel showing the generation of splicing intermediates and products after incubation of radioactively labeled actin pre-mRNA in mock-depleted (lane 3) or U4-depleted (lanes 4–8) extract preincubated with increasing amounts of unlabeled actin pre-mRNA. Lanes 1 and 2 are standard splicing controls incubated for 0 or 30 min, respectively. Lanes 3–8 are the same reactions as in panel B. Bands corresponding to splicing substrate, intermediates, and products are indicated to the left of the gel (black and white boxes, 5′ and 3′ exons; black line, intron). The absolute splicing efficiency of each reaction is indicated below the gel.
To investigate the domain structure and interactions of yeast U4, we have developed an assay for in vitro reconstitution of functional U4 snRNPs. By using DNA oligonucleotide-targeted degradation by RNase H, extract was specifically depleted of endogenous U4, abolishing splicing. Subsequent addition of in vitro transcribed (IVT) U4 snRNA restored up to 50% of the splicing activity of the extract. By using this system, we were able to define the minimum functional sequence of yeast U4 snRNA. Our results reveal that in contrast to mammalian U4, the 3′ stem–loop of yeast U4 is essential for interaction with U6 and thus for splicing. Strikingly, a number of U4 mutants base-paired to U6 more efficiently than wild-type U4. The observation that these mutants were nevertheless impaired in their ability to support splicing demonstrates that U4 plays an important role subsequent to U4/U6 di-snRNP formation.
RESULTS
The 5′ end of U4 snRNA is efficiently degraded under conditions of active splicing
To develop an in vitro assay for reconstitution of functional yeast U4 snRNPs, we needed to inactivate the endogenous U4 and thereby block the splicing activity of the extract. An effective technique for depletion of specific RNA molecules from extract is oligonucleotide-targeted degradation by RNase H. A previous attempt to develop a yeast U4 in vitro reconstitution assay used a complementary DNA oligonucleotide to direct RNase H degradation of U4 nucleotides 72–92 but was unsuccessful (Horowitz and Abelson 1993). This oligonucleotide had been shown to direct nearly complete degradation of the target sequence (Xu et al. 1990); however, degradation of this central region of U4 failed to abolish splicing activity (Horowitz and Abelson 1993), presumably because a functional portion of U4 remained intact. Here, we instead targeted the 5′ end of U4 (Fig. 1A), as it has been shown to be essential for interaction with U6 and, consequently, for splicing (Vankan et al. 1990; Wersig and Bindereif 1990, 1992).
Normally, the 5′ end of U4 is found base-paired to U6, forming intermolecular stem II (Fig. 1A), and, as a result, is almost completely resistant to oligonucleotide-targeted RNase H degradation (Xu et al. 1990). To circumvent the protection provided by U4/U6 stem II and to accomplish efficient U4 degradation, we took advantage of the changes in the U4 secondary structure that take place during the splicing cycle. For the active spliceosome to be formed, base-pairing interactions between U4 and U6 must be broken, resulting in the release of U4 from the spliceosome in the form of a free particle. In this U4 species, the 5′ region normally engaged in base-pairing with U6 is believed to adopt a largely single-stranded conformation that should be more accessible to the targeting DNA oligonucleotide (Fig. 1A). To produce conditions of active splicing, unlabeled actin pre-mRNA was added to yeast splicing extract in the presence of ATP. Titration of actin pre-mRNA showed that increasing amounts of pre-mRNA allowed higher U4 degradation efficiencies, with 80 fmol pre-mRNA resulting in nearly complete depletion of full-length U4 (Fig. 1B). The amount of actin pre-mRNA necessary for efficient depletion of U4 was found to be extract-dependent, ranging from 15–80 fmol. Under these conditions, U4 snRNA is specifically depleted, and there is no effect on the other snRNAs (Fig. 1C). The remaining U4 fragment (nucleotides [nt] 31–160, labeled U4-RNaseH) is visible but is substantially degraded.
Degradation of the 5′ end of U4 snRNA inhibits splicing in vitro
A critical requirement for the development of a useful U4 in vitro reconstitution assay is that degradation of endogenous U4 must inhibit the splicing activity of the extract. To test the effectiveness of splicing inhibition, U4-depleted extract was incubated with radioactively labeled pre-mRNA, followed by gel electrophoresis to separate the splicing substrate from any intermediates or products formed. Near-complete depletion of the full-length U4 resulted in complete inhibition of splicing in the extract relative to the mock-depleted control (Fig. 1D, cf. lanes 3 and 8). Given that the number of splicing cycles that can be performed by a specific extract is limited (Raghunathan and Guthrie 1998a), it was important to ensure that the splicing performed during the degradation of U4 was not compromising the splicing potential of the extract. Incubation of mock-depleted extract in the presence of 80 fmol pre-mRNA decreased the subsequent splicing activity of the extract by ∼20% compared with mock-depleted extract incubated in the absence of pre-mRNA (cf. lanes 2 and 3). Mock-depleted extract was generally able to splice ∼40%–70% of the labeled actin pre-mRNA, while depletion of U4 consistently reduced this number to <1%.
IVT U4 RNA restores splicing in U4-depleted extract
Having identified conditions that allowed efficient depletion of endogenous full-length U4 and concomitant inhibition of splicing, we attempted to restore splicing activity by addition of IVT U4. Titration of IVT U4, from 28–2830 nM, showed effective restoration of splicing relative to depleted extract (Fig. 2A, cf. lanes 3–9 to lane 2). Maximum reconstitution was achieved by addition of 283 nM IVT U4 (lane 6). This U4 concentration corresponds to about 300-fold more than endogenous U4 levels (see Discussion) (Brenner and Guthrie 2006). Addition of >283 nM IVT U4 resulted in decreased splicing; therefore, subsequent experiments used 283 nM U4.
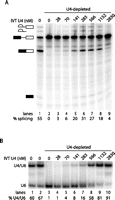
Reconstitution of splicing activity in U4-depleted extract by IVT U4 is not limited by the extent of base-paired U4/U6 formation. (A) Maximum splicing reconstitution is obtained with addition of 283 nM IVT U4. Denaturing splicing gel as in Figure 1D. (Lane 1) Mock-depleted extract. (Lanes 2–9) Depleted extract with IVT U4 added as indicated above lanes. Absolute splicing efficiency of each reaction is indicated below gel. (B) Extent of base-pairing with U6 snRNA increases with added IVT U4. Non-denaturing solution hybridization gel probed using a radioactively labeled oligonucleotide complementary to U6, showing the base-pairing status of U6. (Lane 1) Untreated splicing extract. (Lane 2) Mock-depleted extract. (Lanes 3–10) U4-depleted splicing extract with IVT U4 added as indicated. The percentage of U6 in base-paired complex is shown below the gel.
A range of degradation oligonucleotide concentrations, from 0.05–50 μM, was examined to identify the optimal concentration for depletion of full-length U4 while maintaining the ability of the depleted extract to restore splicing upon addition of IVT U4. This analysis showed that maximum reconstitution was achieved after depletion with 0.8 μM oligonucleotide (data not shown).
To ascertain whether U4/U6 di-snRNP levels were limiting for splicing reconstitution, we measured the corresponding U4/U6 levels for each of the splicing reactions in Figure 2A (Fig. 2B). In contrast to the splicing efficiency, which peaked at 283 nM U4, U4/U6 levels continued to increase with increasing U4 to a maximum of 91% at 2830 nM (lane 10). In untreated splicing extract, 60% of U6 was base-paired (lane 1) compared with 67% in mock-depleted extract (lane 2). By comparison, only 16% of U6 was base-paired when we reconstituted with 283 nM U4 (lane 7), suggesting that there was insufficient di-snRNP to support maximal splicing. Nevertheless, when 566 nM U4 was added, yielding 58% base-pairing of U6 (lane 8), the splicing efficiency decreased (Fig. 2A, lane 7). Further increases in U4 concentration resulted in further decreased splicing efficiency.
Splicing activities of U4 3′ truncation mutants
To examine the functional domains of yeast U4, we first determined the minimum sequence that supports U4/U6 formation and splicing using our reconstitution assay with U4 mutants. Given that stems I and II and the 5′ stem–loop are all required for efficient spliceosome assembly and splicing in human extract and Xenopus oocytes (Vankan et al. 1990; Wersig and Bindereif 1990, 1992), we tested a mutant containing these three structural features, which consisted of nt 1–68. This mutant, U4 1–68, lacks the Sm binding site, the 3′ stem–loop, and most of the central domain (Fig. 3A); however, it does contain the 5′ portion of the central domain identified by Wersig and Bindereif (1992) and Vankan et al. (1992) as being important for splicing activity.
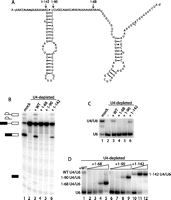
The 3′ stem–loop of U4 is required for efficient base-pairing to U6. (A) Secondary structure of U4 snRNA with the 3′ ends of the truncation mutants indicated. (B) U4 truncations lacking the 3′ stem–loop do not support reconstitution of splicing. Denaturing gel analysis of splicing in mock-depleted (lane 1) or U4-depleted (lanes 2–6) extract reconstituted using wild-type (WT) or mutant IVT U4. Splicing substrate, intermediates, and products are indicated to the left of the gel, as in Figure 1. (C) The 3′ stem–loop of U4 is required for base-pairing to U6 at 283 nM U4. Non-denaturing solution hybridization gel analysis of the base-pairing status of U6 in mock-depleted (lane 1) or U4-depleted (lanes 2–6) extract reconstituted using WT or mutant IVT U4. (D) U4 truncations can be driven into complexes with U6 by increasing their concentration above 283 nM. Non-denaturing solution hybridization analysis of extent of U4/U6 formation as in panel C. (Lane 1) 283 nM full-length IVT U4. Increasing concentrations of truncated U4 mutants: 283, 566, 1132, and 2830 nM (1–142 only to 1132 nM).
To examine the effect of truncating U4 at nt 68, U4 1–68 transcript was added to extract depleted of endogenous U4, and its ability to restore splicing was assessed. Unexpectedly, the addition of U4 1–68 to the U4-depleted extract did not result in reconstitution of splicing activity (Fig. 3B, lane 4), in contrast to the ability of wild-type IVT U4 to restore splicing to 21% from 1% in the U4-depleted extract (Fig. 3B, lanes 2 and 3; splicing and U4/U6 formation efficiencies reported in the text are averages from at least three measurements; see Table 1).
Di-snRNP formation and splicing reconstitution efficiencies of the U4 mutants
Lack of splicing reconstitution by U4 1–68 may be due to the absence of the 3′ portion of the central domain (nt 69–90). While not essential for splicing, deletion of this sequence from mammalian U4 has an appreciable effect on splicing efficiency (Wersig and Bindereif 1992). We therefore constructed a longer 3′ truncation mutant containing the entire central domain (U4 1–90) (Fig. 3A) and examined its ability to reconstitute U4-depleted extract. Surprisingly, this mutant was also unable to reconstitute splicing (Fig. 3B, lane 5).
The final U4 3′ truncation mutant we examined, U4 1–142, contains the 3′ stem–loop in addition to the complete central domain, stems I and II, and the 5′ stem–loop and thus lacks only the 3′ terminal domain of U4, which contains the Sm protein binding site (Fig. 3A). Consistent with reconstitution studies in human nuclear extract (Wersig and Bindereif 1992), we found that the 3′ terminal domain of yeast U4 was not essential for splicing in vitro (Fig. 3B, lane 6), although the average level of splicing restored, determined from multiple measurements, was only 49% of that restored by wild-type U4 (Table 1).
The lack of splicing reconstitution by U4 1–68 and 1–90 could be due to the removal of a functionally essential region of the molecule or to decreased stability of these mutants in splicing extract. To investigate this second possibility, we examined the stability of wild-type and mutant IVT U4 snRNA after incubation in U4-depleted splicing extract. The three mutants were all less stable than wild-type IVT U4, with U4 1–142, which was active in the reconstitution assay, having an intermediate stability (Table 1). This suggests that the inability of the shorter 3′ truncation mutants to function in splicing is not due to a lack of stability; rather, these mutants lack a functionally essential component of U4.
Interaction of the U4 3′ truncation mutants with U6
Despite the fact that U4 truncation mutants 1–68 and 1–90 contain the only regions of U4 known to interact with U6, their inability to reconstitute splicing in U4-depleted extract could be caused by reduced or absent base-pairing with U6 snRNA. To investigate this possibility, we measured the base-pairing status of U6. As expected, U4 depletion produced extract in which U6 was found exclusively in the free snRNP (Fig. 3C, lane 2). Both wild-type U4 and U4 1–142 were able to reconstitute formation of the di-snRNP in U4-depleted extract to ∼20% of normal levels (Fig. 3C, lanes 3 and 6); however, in the majority of experiments we found that U4 1–142 yielded more U4/U6 than the wild type (Table 1). U4 1–68 and 1–90 were unable to reconstitute di-snRNP formation at the concentration tested (Fig. 3C, lanes 4 and 5).
The reduced ability of U4 truncations to form U4/U6 may be due to a kinetic defect in base pair formation or, alternatively, to reduced stability of the base-paired product. To test the first possibility, we allowed the reconstitution reactions to proceed for varying lengths of time up to 36 min. We found that the fraction of U4/U6 did not change after 12 min, indicating that the reactions had reached equilibrium (data not shown). To test the second possibility, we performed titrations of the truncated U4 mutants, from 141 to 2830 nM, and subsequently examined U4/U6 levels. When we fit a binding curve to the titration data to calculate an apparent KD (see Materials and Methods), we found a significant difference between the mutants, ranging from 1528 nM for 1–68 to 670 nM for 1–142, compared with 800 nM for the wild type (Table 1). These results are consistent with the truncations affecting di-snRNP stability. Surprisingly, the 1–142 mutant appears to have a more stable interaction with U6 than wild-type U4, suggesting that the Sm ring of U4 may antagonize base-pairing with U6.
The ability of U4 1–68 and 1–90 to base pair to U6 at high concentration suggested that they may be able to reconstitute splicing at these concentrations. We therefore tested splicing reconstitution using these mutants at concentrations from 283–2830 nM, which corresponded to U4/U6 levels from 0%–98% for 1–68 and 0%–72% for 1–90 (Fig. 3D, lanes 2–9). Neither mutant was able to reconstitute splicing at any concentration tested (data not shown). U4 1–142 was similarly tested in a splicing reconstitution titration assay and, like the wild type, showed maximum splicing at 283 nM, despite increasing di-snRNP formation up to the highest concentration tested (1132 nM, yielding 93% di-snRNP; lane 12).
We conclude that the inability of U4 1–68 and 1–90 to reconstitute splicing does not stem exclusively from their reduced ability to interact with U6, as they do not support splicing even under conditions where high levels of U4/U6 are formed. Intriguingly, these experiments point to the 3′ stem–loop of U4 as an important determinant of U4/U6 di-snRNP formation.
Mutational analysis of the 3′ stem–loop of U4 snRNA
To directly test whether the 3′ stem–loop is required for di-snRNP formation, we constructed a mutant, Δ3′SL, that was missing the entire 3′ stem–loop (nt 91–141) (see Fig. 3A) but still contained the Sm binding site. Consistent with the above experiments, this mutant was unable to participate in di-snRNP formation at the normal concentration (Fig. 4B, lane 4) and was also unable to restore splicing at this concentration or any other up to 2830 nM (Fig. 4C, lane 4; data not shown). Indeed, Δ3′SL had the highest KDapp of any U4 mutant tested, 2192 nM (Table 1).
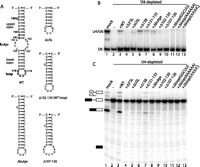
U4 3′ stem–loop mutations that form comparable levels of U4/U6 to the wild type (WT) are nevertheless defective in splicing reconstitution. (A) Secondary structures of constructs used in this experiment: WT U4 3′ stem–loop, deletion of the 11-nt bulge (Δbulge), the upper stem and bulge (ΔUSL), the bulge and lower stem (Δ102–130 [WT loop]), or the lower stem and loop (Δ107–126). Locations of deleted (138; 131–133) or mutated (loop mutations) nucleotides are shown in bold on the WT stem–loop. (B) Deletions or mutations at many positions in U4's 3′ stem–loop affect U4/U6 formation. Non-denaturing gel showing the base-pairing status of U6 in mock-depleted (lane 1) or U4-depleted (lanes 2–13) extract reconstituted using WT or mutant IVT U4. The positions of free U6 and the U4/U6 di-snRNA are indicated to the left of the gel. (C) Deletions and mutations in U4's 3′ stem–loop reduce splicing reconstitution. Denaturing gel showing splicing activity in mock-depleted (lane 1) or U4-depleted (lanes 2–13) extract reconstituted using WT or mutant IVT U4 as in B. Splicing substrate, intermediates, and products are indicated to the left of the gel, as in Figure 1.
Given the unexpected importance of the 3′ SL for di-snRNP formation and splicing, we undertook a more detailed mutational analysis to identify functionally important regions within this domain. We constructed 10 U4 mutants containing deletions or base mutations throughout the 3′ stem–loop (Fig. 4A) and analyzed the ability of each to restore splicing and di-snRNP formation in U4-depleted extract.
To investigate whether the nucleotides important for di-snRNP formation are located in the upper portion of the 3′ stem–loop, we examined the effects of a number of deletions in this region. We constructed a mutant, ΔUSL, in which the entire upper stem and central bulge were deleted, leaving only the distal portion of the stem and loop (Fig. 4A). While this mutant had a KDapp of 1137 nM for di-snRNP formation, the splicing activity restored was <10% that of the wild type (Fig. 4B and C, lane 5; Table 1), indicating that sequences important for both di-snRNP formation and splicing had been deleted. To identify these sequences, we constructed three mutants containing smaller deletions within the upper portion of the 3′ stem–loop. Deletion of U138, a highly conserved bulged nucleotide (Myslinski and Branlant 1991), resulted in better-than-wild-type interaction with U6 (KDapp = 650 nM) but restored only 10% of the splicing activity seen with wild-type U4 (lane 6; Table 1). In contrast, deletion of nucleotides 131–133, a mutation previously shown to produce a cold-sensitive phenotype in vivo (Hu et al. 1995b), resulted in levels of di-snRNP formation slightly below that of the wild type (836 nM) and splicing levels that were 28% that of the wild type (lane 7; Table 1). Finally, similar to Δ138, deletion of the 9-nt central bulge of the 3′ stem–loop, Δbulge (Fig. 4A), resulted in above-wild-type interaction with U6 (762 nM) but substantially below-wild-type restoration of splicing (11% of the wild type) (lane 8; Table 1).
While deletions within the upper portion of the 3′ stem–loop identified nucleotides that contribute to splicing, none of these mutations resulted in the strong block to di-snRNP formation that was produced by deletion of the entire 3′ stem–loop. To investigate whether a sequence essential for di-snRNP formation resides within the lower portion of the 3′ stem–loop, we generated five mutants containing deletions within this region. Deletion of the central bulge and distal portion of the stem, leaving the two sides of the upper stem connected by the wild-type loop sequence, produced Δ102–130 (Fig. 4A). This mutant had an apparent KD for di-snRNP formation of 1105 nM and low, but above background, splicing (Fig. 4B,C lane 9; Table 1). Deletion of the entire lower stem and loop, Δ107–126 (Fig. 4A), severely impaired di-snRNP formation (KDapp, 1666 nM) and splicing (lane 10; Table 1), indicating that a sequence essential for di-snRNP formation had been deleted.
The observation that the apparent KD for U4/U6 formation of Δ102–130 was substantially lower than Δ107–126 suggested that the loop nucleotides of the 3′ stem–loop are essential for di-snRNP formation. To test this hypothesis, we generated mutants in which the 6 nt of the loop were replaced. First, we asked whether the sequence of the loop was important by replacing the wild-type 3′-UUUUCG-5′ sequence with all As. This mutant formed no U4/U6 di-snRNP at the standard concentration and, consequently, did not support splicing (Fig. 4B,C, lane 12), indicating that nucleotide identity in the loop is important for U4/U6 formation. An alternative loop sequence from Trypanosoma brucei had previously been shown to form U4/U6 in vivo in the context of a chimeric U4 (Bordonné et al. 1990). We tested this loop sequence, 3′-GGCUU-5′, and found that, as with the all-adenosine loop, it supported neither base pair formation nor splicing (lane 11). Finally, to test whether the length of the loop was important, we made a version lacking G114 at the 5′ end (3′-UUUUC-5′). Similarly to the other loop mutants, this version of U4 was inactive in base pair formation and splicing (lane 13). Although all of the 3′ stem–loop mutants were able to base pair with U6 at concentrations >283 nM, those that were unable to form di-snRNP or splice at 283 nM were also unable to splice at higher concentrations (i.e., Δ3′SL, Δ107–126, and all loop mutants; data not shown). Based on these experiments, the loop appears to be the most important portion of the 3′ stem–loop for both U4/U6 formation and splicing.
Function of the central domain
Having demonstrated that the 3′ stem–loop of U4 was required for productive U4/U6 formation (i.e., di-snRNP formation that supports splicing), we sought to determine whether it might also have a function later in the splicing reaction. We attempted to remove the 3′ portion of U4 after di-snRNP formation using an oligonucleotide targeting the central domain (Fig. 5A). This resulted in separate but stable 5′ and 3′ portions of U4 (data not shown). Degradation was performed under either splicing conditions (in the presence of added pre-mRNA) or nonsplicing conditions (in the absence of added pre-mRNA as done previously) (Horowitz and Abelson 1993).
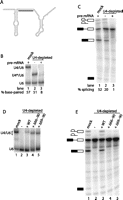
The central domain of U4 functions during U4/U6 formation and splicing. (A) Secondary structure of U4 (gray) with targeting oligonucleotide binding site indicated (black line). (B) U4 requires its central domain to re-associate with U6. Non-denaturing solution hybridization gel showing the base-pairing status of U6 in mock-depleted extract (lane 1) or extract depleted of U4 in the absence (lane 2) or presence (lane 3) of added unlabeled actin pre-mRNA. The positions of base-paired wild-type (WT) U4/U6, U6 base-paired to the truncated 5′ portion of U4 (U4*/U6), and free U6 are indicated to the left of the gel. The fraction of U6 base-paired to U4 in each reaction is indicated below the gel. (C) Degraded U4 supports splicing when associated with U6. Denaturing gel showing splicing activity in mock-depleted extract (lane 1) or extract depleted of U4 in the absence (lane 2) or presence (lane 3) of pre-mRNA added during the U4 degradation step. Splicing substrate, intermediates, and products are indicated to the left of the gel, as in Figure 1. Absolute splicing efficiency of each reaction is indicated below the gel. (D) Deletion of U4's central domain increases steady-state levels of U4/U6 di-snRNP. U4/U6 analysis as in B of mock-depleted (lane 1) or depleted (lanes 2–5) extracts reconstituted with no U4 (lane 2) or U4 variants as shown. Note that U4/U6 migrates faster with the mutant U4s due to their smaller size. (E) U4's central domain is necessary for splicing. Splicing analysis of the reactions in panel D.
In the absence of targeting oligonucleotide (Fig. 5B, lane 1), the majority of U6 is associated with U4. Similarly, following cleavage of U4 under nonsplicing conditions (lane 2), the majority of U6 remained associated with the 5′ portion of U4 (containing nt 1–64), demonstrating that the complex of U6 with truncated U4 was stable. Cleavage of U4 under splicing conditions, however, resulted in the conversion of almost all U6 to the free snRNA (lane 3). We infer that the truncated U4 and U6 were separated during splicing and were unable to re-anneal. Surprisingly, extract containing the cleaved U4/U6 complex supported subsequent splicing of radioactively labeled pre-mRNA at ∼40% of normal splicing levels (Fig. 5C, lanes 1,2), while extract containing truncated U4 separated from U6 was not able to support splicing (lane 3). These results demonstrate that the 5′ portion of U4 was able to remain in a stable, functional complex with U6 after central domain degradation and that the 3′ stem–loop does not need to be physically attached to the 5′ end of U4 (nt 1–64 in this experiment) after U4/U6 formation. Due to the stability of the 3′ end of U4, these experiments were unable to determine the step at which splicing is blocked in the absence of the 3′ stem–loop.
These results raise the possibility that U4's central domain (Figs. 1A, 3A) is not necessary for U4 function beyond its role in joining the two functionally essential ends of the molecule. To test this, we reconstituted splicing with two U4 mutants, Δ66–90 and Δ69–90, in which the central domain was deleted. Wersig and Bindereif (1992) previously showed that the nucleotides in human U4 corresponding to yeast positions 65–68 contributed to splicing in vitro, so we made one deletion that removed these nucleotides along with the rest of the central domain (Δ66–90), and another (Δ69–90) that retained them. Surprisingly, both mutants base-paired with U6 far more than wild-type U4 (Fig. 5D, lanes 3–5). Titrations yielded KDapp values of 390 and 470 nM, respectively, compared with 800 nM for the wild type (Table 1), the most stable interactions with U6 of any U4 mutants that we have tested. When the ability of these mutants to support splicing was measured, however, Δ66–90 had no activity and Δ69–90 had very little activity, albeit consistently above depleted extract (Fig. 5E; Table 1), consistent with the observations of Wersig and Bindereif (1992). In contrast to the oligo degradation experiments (Fig. 5B,C), these deletion experiments suggest that the central domain contributes to splicing but antagonizes U4/U6 formation (see Discussion).
snRNP analysis of U4 tri-snRNP formation
Certain U4 mutants described above were notable for having a lower apparent KD than wild-type U4 (U4 1–142, Δ138, Δbulge, Δ66–90, and Δ69–90), yet none of them supported splicing as well as the wild type. One possibility is that these U4 variants were defective in the next assembly step after U4/U6 di-snRNP formation, namely, U4/U6.U5 tri-snRNP formation. To test this, we measured the ability of these mutants to assemble into tri-snRNPs at several concentrations (Fig. 6).
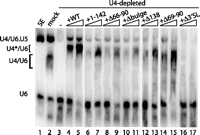
U4 deletion mutants are defective in triple-snRNP formation. Non-denaturing Northern blot analysis of unextracted (i.e., protein-containing) samples. Untreated extract (lane 1), mock-depleted extract (lane 2), and depleted extract reconstituted with nothing (lane 3) or with IVT U4 variants (283 or 566 nM, lanes 4–17). Location of snRNP species indicated by labels at left. U4*/U6 indicates aberrantly migrating U4/U6 di-snRNP seen in all reconstituted extracts.
SnRNP levels in extract can be analyzed on a non-denaturing gel without phenol extracting the samples (i.e., with proteins present) (Cheng and Abelson 1987; Raghunathan and Guthrie 1998a). Under these conditions, all of the base-paired U6 is actually in tri-snRNP, and there is no U4/U6 di-snRNP present (Fig. 6, lane 1; Raghunathan and Guthrie 1998a). Upon addition of ATP, tri-snRNP dissociates into free U4 and U6 snRNPs, which subsequently reform U4/U6 and tri-snRNP (lane 2). Depletion of U4 from extract leads to the complete absence of U4/U6 and a strongly reduced signal at the position where tri-snRNP runs (lane 3). The residual signal at the tri-snRNP position may reflect incomplete degradation of U4 or may simply be nonspecific background; but in any case, there is no detectable splicing under these conditions (see Fig. 1D).
All of the U4 mutants tested yielded less tri-snRNP after reconstitution than did wild-type U4, which reconstituted 24% of the level of tri-snRNP seen in mock-depleted extract. In particular, the five U4 mutants that had lower apparent KDs than the wild type, namely, 1–142, Δ66–90, Δbulge, Δ138, and Δ69–90 (lanes 6–15), had 16%–27% as much tri-snRNP as the wild type yet more than Δ3′SL, which was comparable to the depleted lane (cf. lane 3 to lanes 16,17). The amount of tri-snRNP formed did not increase with additional U4 above 283 nM, in contrast to di-snRNP formation (e.g., cf. lanes 4 and 5), which is consistent with the maximum splicing activity occurring at 283 nM, and may suggest that splicing is limited by tri-snRNP availability.
This demonstrates that all of these mutants are defective in tri-snRNP formation. Notably, they support a wide range of splicing efficiencies, from 49% of the wild type for U4 1–142% to 0% for Δ66–90. The lack of correlation between tri-snRNP levels and splicing activity indicates that there is yet another step after tri-snRNP formation in which the 3′ stem–loop and central domain of U4 play a role (see Discussion).
An important observation is that the di-snRNP present in the reconstituted lanes of the gel in Figure 6 (lanes 4–17) has a lower mobility than di-snRNP from mock-depleted extract (lane 2). We have confirmed that this band, labeled U4*/U6 in the figure, does not contain U5 snRNA (data not shown), so the aberrant mobility must be either due to a difference in the protein complement compared to untreated extract or due to a conformational change in one or more components (most likely U4). This may in turn be responsible for the reduced levels of tri-snRNP and splicing activity upon reconstitution.
Oligonucleotide accessibility of the 3′ stem–loop of U4 snRNA
Mutation of specific regions of the 3′ stem–loop of U4 may inhibit di-snRNP formation or subsequent splicing by destabilizing an interaction with a protein. To investigate whether the 3′ stem–loop participates in protein/RNA interactions, we examined the ability of oligonucleotides complementary to the 5′ or 3′ side of this domain (Fig. 7A) to direct degradation of U4 in extract by RNase H in the presence or absence of proteins. Surprisingly, the two sides of this domain exhibited unequal oligonucleotide accessibilities. Only the 5′ side was efficiently degraded in the presence of proteins (Fig. 7B, lanes 2,3), and neither side was susceptible to degradation in the absence of proteins (Fig. 7B, lanes 6,7). Because the Sm site of U4 is known to bind the Sm proteins, this region was used as a degradation control. As expected, it was protected from degradation in the presence of proteins but was accessible to the oligonucleotide, and therefore susceptible to degradation, in the absence proteins (Fig. 7B, lanes 4,8). These results suggest that the 3′ stem–loop of U4 interacts with at least one protein.
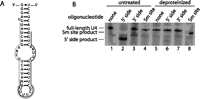
Proteins confer asymmetric accessibility to U4's 3′ stem–loop in untreated splicing extract. (A) Secondary structure of the U4 3′ stem–loop showing the binding sites of oligonucleotides targeting the 5′ and 3′ sides (black and gray lines, respectively). (B) The U4 3′ stem–loop is only degraded in the presence of proteins. Denaturing Northern blot probed for U4, showing U4 size in the absence (lanes 1,5) or presence (lanes 2–4,6–8) of oligonucleotide targeting the 5′ or 3′ side of the 3′ stem–loop, or the Sm site. Lanes 1–4 are in the presence of proteins, while lanes 5–8 are in the absence of proteins.
DISCUSSION
Conditions for successful U4 reconstitution
In this study, we have developed the first in vitro splicing reconstitution system for U4 snRNP in yeast. A key requirement of this assay was to deplete endogenous U4 snRNA under conditions of active splicing so that the 5′ end of U4 targeted for RNase H degradation was no longer base-paired to U6 and was therefore accessible to the targeting DNA oligonucleotide. We were able to reconstitute the splicing activity of the depleted extract by addition of IVT U4, which allowed us to discover an unexpected role for the 3′ stem–loop in di-snRNP formation, as well as in later steps of spliceosome assembly (steps at which U4 3′ domains may function are summarized in Fig. 8).
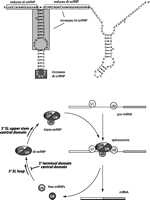
Model of functional domains in the 3′ portion of U4 and the spliceosome assembly steps at which they function. The region of U4 that promotes di-snRNP formation is indicated in black, the region that increases tri-snRNP formation is in dark gray, the region that decreases di-snRNP levels is boxed in white, and the region that both decreases di-snRNP and increases tri-snRNP is boxed in light gray. In the lower panel, the portions of U4 tested in this work that we propose to be functionally important are shown in bold, with arrows indicating the points in the splicing cycle at which they act.
The amount of splicing reconstitution we observed was somewhat lower than that seen with other snRNAs (our range was 21%–76% of the mock reaction, compared with 50%–100% of untreated extract for U6 [Fabrizio et al. 1989], and 55%–70% of the mock reaction for U2 [McPheeters et al. 1989]), and required higher concentrations of IVT U4 (283 nM compared to 40–125 nM for U6 and U2 [Fabrizio et al. 1989; McPheeters et al. 1989; McGrail et al. 2006] and 125 nM for U5 [McGrail and O'Keefe 2008]). Approximately 50% of the added IVT U4 was degraded in extract over the course of splicing reconstitution. In addition, the aberrant mobility of the reconstituted di-snRNP on a non-denaturing gel (Fig. 6) suggests that much of the IVT U4 is either misfolded or does not associate properly with proteins. Nevertheless, its ability to reconstitute tri-snRNP with normal mobility, as well as splicing activity, suggests that some fraction of the U4 interacts normally with the rest of the splicing machinery.
The decrease in reconstitution efficiency seen with IVT U4 concentrations >283 nM may be due to increased competition for U4-specific proteins, effectively diluting the proteins available such that fewer U4 RNA molecules receive the full protein complement required for functionality. Alternatively, an inhibitory component in the IVT solution (such as residual acrylamide) could be responsible for the inhibition.
Conditions for complete depletion of endogenous U4 from extracts had to be determined for each extract tested, requiring 15–80 fmol of actin, compared with 4 fmol for a standard splicing reaction. This may reflect the need to cycle the entire pool of endogenous U4 through the splicing pathway, perhaps multiple times, to ensure complete degradation. We cannot explain the variability between extracts. It was also surprising that U4 depletion was only successful under conditions of active splicing, as ATP alone is known to cause the dissociation of tri-snRNP in yeast as well as human extract (Berget and Robberson 1986; Raghunathan and Guthrie 1998b). This suggests the possibility that U4 released from the spliceosome is in a different conformation from U4 released from tri-snRNP under nonsplicing conditions, with the 5′ end of the former being more accessible than the latter. Another possibility is that the difference in oligo accessibility is due to differences in protein composition of the two types of free U4 snRNP.
The 3′ stem–loop of yeast U4 is required for efficient di-snRNP formation
Reconstitution by U4 3′ truncation mutants showed that the 3′ stem–loop plays a critical role in promoting the interaction with U6 that is required for subsequent reconstitution of splicing. This was unexpected, as a number of previous mutational analyses of U4 in human nuclear extract and Xenopus oocytes have shown that deletion of this domain does not significantly inhibit di-snRNP formation (Vankan et al. 1990; 1992; Wersig and Bindereif 1990; 1992). Indeed, these studies further demonstrated this domain to be dispensable for spliceosome assembly and even for splicing, although efficiency was reduced compared with wild-type U4. Similarly, a series of point mutations throughout the 3′ stem–loop of yeast U4 were found to be functional in vivo, although deletion of nt 131–133 did result in a cold-sensitive phenotype (Hu et al. 1995b).
In contrast to these studies suggesting that the 3′ stem–loop is not a functionally important element of U4 snRNA, an in vivo analysis by Bordonné et al. (1990) found that substitution of the yeast U4 3′ stem–loop with the comparable region of T. brucei U4, an 11-nt stem–loop, decreased interaction with U6 by 60%–70% and was lethal. Similarly, the 3′ stem–loop of human U4atac snRNA has been shown to be required for in vivo activity (Shukla et al. 2002). Hence, the requirement for the U4 3′ stem–loop appears to vary between organisms and even between the major and minor spliceosome. Furthermore, while the 3′ stem–loop of yeast U4 is clearly necessary for di-snRNP formation, there appears to be considerable flexibility in the sequence requirements of this domain.
The U4 3′ stem–loop mutants can be divided into two categories based on their di-snRNP formation and splicing reconstitution efficiencies: those that restored reduced levels of di-snRNP and splicing, and those that restored increased levels of di-snRNP but reduced levels of splicing. We note that the two classes of 3′ stem–loop mutants appear to segregate by location in the stem–loop: Those that increase the apparent KD are in the lower stem–loop, while those that have no significant effect (Δ131–133) or reduce the apparent KD (Δ138 and Δbulge) are in the upper stem.
Two lines of evidence demonstrate the importance of the loop nucleotides of U4's 3′ stem–loop (nt 114–119) in di-snRNP formation. First, deletion of either the upper stem (ΔUSL) or the lower stem (Δ102–130) has only a modest effect on the apparent KD for U4/U6 formation. Importantly, neither of these changes the loop sequence. In contrast, Δ107–126, which removes the loop along with the lower stem, dramatically increases the apparent KD. Second, mutations in the loop itself have a greater impact on U4/U6 formation than deletion of either the upper or lower stem, and one of the loop mutants has the second-highest apparent KD of all of the mutations tested, exceeded only by deletion of the entire 3′ stem–loop. Given that di-snRNP must form prior to spliceosome assembly, any mutations that strongly reduce di-snRNP formation will also block splicing. Consistent with this, the only mutations tested that eliminated splicing were those that changed the 3′ loop, with the exception of Δ66–90, which removes central domain nucleotides previously shown to be important for splicing (Wersig and Bindereif 1992).
The loop mutations we have tested indicate that both the length and the sequence of the loop are important. Likewise, the chimeric U4 with an altered loop sequence discussed above had a strong effect on di-snRNP formation in vivo (Bordonné et al. 1990). The even more dramatic reduction in U4/U6 levels seen when we placed this loop sequence (Δloop(GGCUU)) on the U4 wild-type stem may reflect differences between the in vitro and in vivo assays used or may be a consequence of differences in the stem. In contrast, one of the U4 point mutations analyzed in vivo by Hu et al. (1995b), G114U, falls within the loop of the 3′ stem–loop. Alone, this mutation did not produce a phenotype, suggesting either that our deletion of this nucleotide (Δloop(UUUUC)) impacts function more than mutations at this position or that the in vitro assay is more sensitive.
The 3′ stem–loop functions at a late stage of spliceosome assembly
Previous studies of U4 function have mostly focused on its role in di-snRNP formation; however, there is evidence indicating that it plays a role in subsequent steps, such as tri-snRNP formation. In yeast, humans, and Xenopus, deletion of the 5′ stem–loop results in wild-type or better di-snRNP formation but invariably inhibits tri-snRNP formation and splicing (Bordonné et al. 1990; Wersig and Bindereif 1992; Wersig et al. 1992). We also tested the effect of deletion of U4's 5′ stem–loop in our reconstitution system and similarly found increased levels of U4/U6, but no splicing (data not shown). The 5′ stem–loop of U4 is associated with Prp4, which has been implicated in tri-snRNP formation (Bordonné et al. 1990). In addition, mutations in the second WD repeat of Prp4 block dissociation of U4 from the spliceosome, suggesting yet another role for U4 just prior to spliceosome activation (Ayadi et al. 1997).
An unexpected late function (i.e., after di-snRNP formation) of U4's 3′ stem–loop is suggested by the second class of 3′ stem–loop mutants. These mutations, Δ138 and Δbulge, restored similar levels of di-snRNP formation as the wild-type sequence and reduced apparent KDs, yet reduced splicing, indicating that they impair a process subsequent to base-pairing with U6. One possible explanation for the observed elevation of di-snRNP levels is that these mutations strengthen interactions with a splicing factor, such as Prp24, that promotes di-snRNP formation. Such gain-of-function mutations are, however, relatively rare, and it would be surprising to find even one, let alone two, in the relatively limited survey of mutations described here. Alternatively, these mutations may disrupt an interaction that would normally lead to destabilization of U4/U6, namely, unwinding by the Brr2 helicase prior to spliceosome activation. In this case, we would expect to see an accumulation of spliceosomes or multi-snRNP particles (larger than di-snRNP) (Raghunathan and Guthrie 1998b), yet we do not. The remaining possibility is that these residues may play a role in formation or stabilization of the U4/U6.U5 tri-snRNP. We favor this role for the nucleotides on the 3′ side of the upper stem–loop because our analysis of snRNP particles shows an accumulation of di-snRNP and reduced tri-snRNP. One mechanism by which this region of the 3′ stem–loop could promote tri-snRNP formation could be through interaction with a U5 protein.
The 3′ side of the 3′ stem–loop may bind a protein
Our oligonucleotide accessibility data showed strong protein dependence. For an RNA sequence to be degraded by RNase H, it must be available for base-pairing with a complementary DNA oligonucleotide. Binding of this targeting oligonucleotide may be blocked if the RNA sequence is involved in secondary or tertiary structure interactions or in protein binding. In the absence of proteins, the 3′ stem–loop of yeast U4 is expected to adopt a highly stable secondary structure with an estimated melting temperature of 77°C (Owczarzy et al. 2008). Given that the estimated melting temperatures of the oligonucleotides targeting the 5′ and 3′ sides of the 3′ stem–loop are substantially lower (45°C and 43°C, respectively), it is not surprising that they were not able to direct RNase H degradation in the absence of proteins.
The differential accessibility of the two sides of the 3′ stem–loop in the presence of proteins was unexpected. The accessibility of the 5′ side demonstrates that the binding of proteins to U4 changes the secondary structure of the 3′ stem–loop. This may result from a protein binding directly to the 3′ side of the stem, disrupting base-pairing within the stem and thereby protecting the 3′ side while releasing the 5′ side for base-pairing with a targeting oligonucleotide. Notably, a recent crystal structure of the U4 core (the Sm ring with U4's Sm binding site and flanking stem–loops) supports the possibility of a protein binding the protected side of the 3′ stem–loop (Leung et al. 2011). For example, the structure shows that the conserved bulged U in the 3′ stem–loop (U114 in humans, U138 in S. cerevisiae) is positioned away from the Sm ring such that it would be accessible to a protein. The asymmetric pattern of oligonucleotide accessibility is also consistent with the asymmetry of mutational effects observed in previous studies (Hu et al. 1995b).
Role of U4's central domain
As discussed above, our data indicate an unexpected role for the 3′ stem–loop after di-snRNP formation. Surprisingly, the 3′ stem–loop does not have to be connected to the 5′ portion of U4 to carry out this role. Extract in which the central domain of U4 has been completely degraded, leaving stable 5′ and 3′ portions of the snRNA, is still able to support splicing (Fig. 5C; Horowitz and Abelson 1993), implying that once the di-snRNP has formed, separation of the two halves of U4 does not prevent any additional interactions that may occur between them. Surprisingly, deletion of the central domain (nt 66–90 or 69–90) results in higher steady-state levels of U4/U6, suggesting that the central domain antagonizes U4/U6 formation. The higher levels could be due to faster di-snRNP formation, slower tri-snRNP formation, or slower unwinding during spliceosome activation (Fig. 8). Given the accumulation of U4/U6 on snRNP gels and the relatively low levels of tri-snRNP after reconstitution with central domain deletion mutants, we favor the interpretation that the most important function of the central domain occurs during addition of U5 to the U4/U6 di-snRNP. Taken together, these experiments demonstrate that during U4/U6 formation, the central domain is only required to keep the 3′ stem–loop physically attached to U4, while during tri-snRNP formation, the central domain is required to maintain appropriate spacing between the 5′ region and the 3′ stem–loop of U4.
Our results are consistent with previous work on the central domain. In Xenopus, deletion of the central domain had no effect on U4/U6 formation but blocked splicing (Vankan et al. 1990). Genetic experiments in yeast also suggested that the requirements for the central domain are flexible: Deletion of nt 62–88 had only a mild cold-sensitive phenotype (Hu et al. 1995b), and insertion of an extra 76 nt into the central domain had no effect on growth (Hu et al. 1995a).
More detailed analysis of the central domain has shown that only the 5′ end has a strong sequence requirement. In Xenopus, substitution of the first 3 nt (64–66) blocked splicing, but no other 3-nt substitution in the central domain had a strong effect (Vankan et al. 1992). Similarly, small deletions throughout the central domain of human U4 revealed that only the 5′ end (nt 64–67) was important for efficient splicing, whereas other deletions reduced splicing by at most 50% (Wersig and Bindereif 1992).
The ability of the two halves of U4 in the splicing extract to separate, and the inability of the separated halves to reassociate with U6, allows us to estimate the fraction of splicing in a normal assay that occurs in the initial round and the fraction that occurs after recycling of U6 (i.e., dissociation and reassembly) (Raghunathan and Guthrie 1998a,b). The data in Figure 5C demonstrate that separating the 5′ and 3′ portions of U4 reduces splicing by ∼60%, suggesting that the remaining 40% of a standard in vitro splicing yield is catalyzed prior to recycling. Importantly, cleavage of the central domain of U4 thus provides a practical method to study single turnover splicing kinetics.
Role of the 3′ terminal domain
Our titration analysis of U4 allowed us to make quantitative comparisons between the mutants. For example, it is notable that deletion of the 3′ stem–loop gave a higher apparent KD than truncation of U4 at nt 68 (2192 nM vs. 1518 nM), which appears to suggest that removing all of U4 after nt 68 has less impact on U4/U6 formation than simply deleting the 3′ stem–loop. The observation that U4 1–90 has a similar apparent KD to U4 1–68 indicates that the lower KD of U4 1–68, compared with that of Δ3′SL, is not due to the absence of the central domain and must therefore be due to the absence of the 3′ terminal domain (nt 143–160). We conclude that the 3′ terminal domain serves to reduce steady-state levels of U4/U6, because its presence in Δ3′SL increases the apparent KD relative to U4 1–90. Our observation that U4 1–142 has a lower apparent KD than wild-type U4 is consistent with this conclusion.
The function of the 3′ terminal domain in decreasing U4/U6 levels could be due to direct antagonism of the rate of di-snRNP formation or to acceleration of U4/U6 unwinding during spliceosome assembly. Our snRNP gel analysis, which shows no change in tri-snRNP levels between any of the mutants, is more consistent with the former possibility, although it is unclear how the 3′ terminal domain, which includes the Sm protein binding site, would slow di-snRNP formation.
The data presented here reveal the unexpected role played by the 3′ stem–loop of yeast U4 in promoting U4/U6 formation and tri-snRNP assembly, with the loop nucleotides promoting di-snRNP formation and with the upper stem and central domain promoting tri-snRNP formation (Fig. 8). The 3′ terminal domain, which includes the Sm protein binding site, appears to slow the rate of di-snRNP formation. Interestingly, a recent article (He et al. 2011) reports that a mutation in the analogous stem of human U4atac is linked to microcephalic osteodysplastic primordial dwarfism type I (MOPD I), also known as Taybi-Linder Syndrome (TALS). Our data suggest that the U4atac mutation may cause problems in U4/U6 formation or in subsequent tri-snRNP formation. These results emphasize the need for structural information to reveal the nature of the conformational changes that occur during RNA structural rearrangements, such as in the transition from free U4 to the U4/U6 di-snRNP. A thorough understanding of this conformational change will require both static structures as well as information about the dynamic processes that occur during this transition.
MATERIALS AND METHODS
DNA oligonucleotides
U4-targeting oligonucleotides, primers used in site-directed mutagenesis, and U4 and U6 probes are listed in Table 2.
DNA oligonucleotides used in this study
Synthesis of U4 snRNA and actin pre-mRNA
Wild-type U4 was synthesized from StyI-linearized pT7U4 plasmid template (Ghetti et al. 1995). Templates for synthesis of the U4 mutants were generated by site-directed mutagenesis from pT7U4 plasmid. The same forward primer containing the T7 promoter, oligonucleotide A, was used for all mutants, and the reverse primers are listed in Table 2. PCR products were purified by electrophoresis through a denaturing polyacrylamide gel and visualized by ultraviolet shadowing. Following homogenization in a 1.5-mL microcentrifuge tube using a small pestle (Kontes Scientific), the gel slice was mixed with 200 μL dH2O and incubated for 10 min at 70°C. The gel slurry was passed over a DTR column (Edge BioSystems) according to the manufacturer's instructions. DNA was ethanol precipitated and quantified using a NanoDrop spectrophotometer (NanoDrop Technologies). Wild-type U4 and U4 mutants were transcribed in vitro from plasmid or PCR template using the MEGAshortscript Kit (Ambion) according to the manufacturer's instructions. RNA was gel purified and quantified as described for PCR products.
Unlabeled actin pre-mRNA was synthesized from HindIII-linearized JPS149 plasmid (Vijayraghavan et al. 1986; Mayas et al. 2006) using the MEGAshortscript Kit (Ambion) according to the manufacturer's instructions and was gel purified and quantified as described above. Radioactively labeled actin pre-mRNA was transcribed and purified as previously described (Aukema et al. 2009).
Oligonucleotide-directed RNase H degradation of U4
Whole-cell extract was prepared from protease-deficient yeast strain BJ2168 as described (Ansari and Schwer 1995). U4 degradation reactions were performed in 8 μL containing 60 mM KPO4 (pH 7.0), 2.5 mM MgCl2, 3% PEG 8000, 50% (v/v) yeast extract, 10 mM ATP, 0.8 μM targeting oligonucleotide (5′ end or central domain) (Table 2), and 15–80 fmol unlabeled IVT actin pre-mRNA (depending on the splicing extract). No exogenous RNase H was added as yeast extract contains sufficient endogenous RNase H activity. Reactions were incubated for 30 min at 30°C. Oligonucleotide accessibility of the 3′ stem–loop and Sm site was examined as described above except 15 μM targeting oligonucleotide (Table 2) was used. For oligonucleotide accessibility of deproteinized U4, yeast extract was phenol/chloroform extracted, ethanol precipitated, and resuspended in 2× RNase H buffer (40 mM Tris-HCl at pH 7.5, 10 mM MgCl2, 4 mM DTT, 0.012% BSA). Four microliters of this resuspension, containing approximately the amount of U4 found in 4 μL extract, was mixed with targeting oligonucleotide, 2 U RNase H (Ambion), and 10 mM ATP to a volume of 8 μL and incubated as described above.
Northern blot analysis of U4 degradation and IVT U4 stability
U4 degradation reactions were terminated by addition of 200 μL stop buffer (0.3 M sodium acetate, 1 mM EDTA, 0.1% SDS, 34 μg/mL Escherichia coli tRNA). Reactions were phenol/chloroform extracted, ethanol precipitated, and electrophoresed through a 6% (19:1) 7 M urea denaturing polyacrylamide gel in 1× TBE at 400 V for 30 min. RNA was transferred to a nylon membrane (Hybond+, GE Healthcare; Owl semi-dry electroblotter) in 1× TBE for 20 min at 2.5 mA/cm2, which was then probed using a 32P-labeled probe complementary to the 3′ (Fig. 1B,C) or 5′ (Fig. 7B) portion of U4 (Table 2) and exposed to a phosphor screen.
Stability of wild-type and mutant U4 transcripts was tested in the presence of depleted splicing extract. We added 2.4 pmol IVT U4 to a U4 degradation reaction after the 30°C incubation, and it was incubated for 12 min at 23°C. Then 4 fmol cold actin transcript was added and incubated for an additional 30 min at 23°C to simulate the conditions of a splicing reaction. The reactions were then treated as above, and the Northern blot was probed with the 5′ U4 probe (Table 2). For control reactions, 200 μL stop buffer was added to 2.4 pmol of each IVT U4 in the absence of depleted splicing extract and processed as above. Stability was calculated as the percentage of U4 transcript remaining relative to the control.
Autoradiograms were visualized using a Cyclone Phosphorimager and OptiQuant software (Packard Instruments). Image contrast and brightness were linearly adjusted using Adobe Photoshop to enhance clarity.
Pre-mRNA splicing assay
To assess the effect of U4 degradation on splicing activity, 4 fmol internally 32P-GTP-labeled actin pre-mRNA in vitro transcript was added to the degradation reaction and incubated for 30 min at 23°C. To assess reconstitution of splicing in U4-depleted extract by wild-type or mutant U4, 2.4 pmol IVT U4 (unless otherwise indicated) was added following the U4 depletion reaction and incubated for 12 min at 23°C to allow snRNP assembly to occur prior to addition of internally labeled actin pre-mRNA and a further incubation for 30 min at 23°C. Splicing reactions were terminated by addition of 200 μL stop buffer, extracted with phenol/chloroform, ethanol precipitated, and electrophoresed through a 6% (19:1) 7 M urea denaturing polyacrylamide gel for 1 h at 400 V. The gel was then exposed to a phosphor screen at −80°C.
To calculate splicing efficiency, autoradiograms were visualized and quantified as described above. The percentage of splicing was calculated by dividing the intensity of bands corresponding to product (mRNA and lariat) by the total intensity of the starting material plus the product (pre-mRNA, lariat, and mRNA). The level of splicing activity restored by the U4 mutants was expressed as a percentage of that of wild-type U4 by subtracting the residual splicing activity found in the U4-depleted reaction from both the mutant and wild-type U4 activities and dividing the former value by the latter. Splicing efficiencies were determined at least in triplicate.
Analysis of U4/U6 base-pairing
Following the U4 degradation reaction, IVT U4 (2.4 pmol for single reactions, or up to 24 pmol in titrations) was added, and the mixture was incubated for 12 min at 23°C to allow interaction with U6 to occur. Two hundred microliters of stop buffer was then added, and the samples were phenol/chloroform extracted and ethanol precipitated. To examine the effect of degradation of the U4 central domain on U4/U6 interaction, stop buffer was added immediately following the degradation reaction. Precipitated RNA was resuspended in 10 μL hybridization buffer (150 mM NaCl, 50 mM Tris-HCl at pH 7.4, 1 mM EDTA) and incubated for 15 min at 23°C with ∼200–500 fmol (100,000 cpm) 32P-labeled U6–467 probe (Table 2). Samples were electrophoresed through a precooled 9% (29:1) non-denaturing polyacrylamide gel in 1× TBE for 1 h at 4°C at 300 V. The gel was exposed to a phosphor screen at −80°C.
To calculate base-pairing efficiency, autoradiograms were visualized and quantified as described above. Base-pairing efficiency was calculated by dividing the intensity of the U4/U6 band by the total intensity of both bands (U4/U6 and free U6). The level of base-pairing activity restored by the U4 mutants was expressed as a percentage of that of wild-type U4 by subtracting the residual base-pairing found in the U4-depleted reaction from both the mutant and wild-type U4 activities and dividing the former value by the latter. Base-pairing efficiencies were determined at least in triplicate.
Calculation of apparent KD values for U4/U6 formation
Absolute values for di-snRNP formation were plotted as a function of the concentration of added IVT U4 in Kaleidagraph (Synergy Software, version 4.1.2), and the data were fit to the curve U4/U6 (%) = (100 × [U4]2)/([U4]2 + KD2). Values for the error and R2 were calculated in Kaleidagraph. As is frequently found in binding experiments, apparent KD fits were apparently cooperative, and allowing a free parameter for the Hill coefficient yielded a value close to 2. We therefore set the Hill coefficient to exactly 2 for our fits to minimize the number of free parameters. In addition, the apparent KD clearly results from several competing processes, including at least the rate of U4/U6 formation and the rate of Brr2-mediated U4/U6 unwinding. It should not be interpreted as a true equilibrium constant for U4/U6 formation. Nevertheless, the U4 titration data demonstrate that U4/U6 formation behaves as a simple equilibrium, which justifies using the apparent KD to compare the ability of U4 variants to base pair with U6.
SnRNP analysis
To assess the formation of tri-snRNP in depleted extracts reconstituted with U4, 1 mM spermidine was added to the U4/U6 base-pairing reconstitution reaction. Protein/RNA complexes were electrophoresed through a precooled 4% (80:1) native polyacrylamide gel in 1× TGM buffer (50 mM Tris base, 50 mM glycine, 2 mM MgCl2) for 3 h at 4°C at 160 V (Raghunathan and Guthrie 1998a). Gels were transferred to nylon membrane in 20 mM NaPO4 (pH 6.4), 20 min at 2.5 mA/cm2, and then probed with 32P-labeled U6-6D (Table 2).
ACKNOWLEDGMENTS
We thank K. Aukema for technical assistance and H. Oh for initial experiments with loop mutants, as well as other members of the Rader laboratory and Rémy Bordonné for helpful discussions and comments on the manuscript. This work was supported by NSERC Discovery Grant 298521 to S.D.R., NSERC Canada Graduate Scholarship 347734, MSFHR Trainee Award 857(06-1)BM, two UNBC Health Research Project Awards, and a UNBC Research Project Award to A.J.H.
Footnotes
-
↵2 Corresponding author.
E-mail rader{at}unbc.ca.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031757.111.
- Received December 7, 2011.
- Accepted January 26, 2012.
- Copyright © 2012 RNA Society








