
Known and novel post-transcriptional regulatory sequences are conserved across plant families
- 1Department of Biochemistry, Cellular and Molecular Biology, The University of Tennessee, Knoxville, Tennessee 37996, USA
- 2Graduate School of Genome Science and Technology, The University of Tennessee, Knoxville, Tennessee 37996, USA
- 3Dipartimento di Chimica Strutturale e Stereochimica Inorganica, Università degli Studi di Milano, 20133 Milano, Italy
Abstract
The sequence elements that mediate post-transcriptional gene regulation often reside in the 5′ and 3′ untranslated regions (UTRs) of mRNAs. Using six different families of dicotyledonous plants, we developed a comparative transcriptomics pipeline for the identification and annotation of deeply conserved regulatory sequences in the 5′ and 3′ UTRs. Our approach was robust to confounding effects of poor UTR alignability and rampant paralogy in plants. In the 3′ UTR, motifs resembling PUMILIO-binding sites form a prominent group of conserved motifs. Additionally, Expansins, one of the few plant mRNA families known to be localized to specific subcellular sites, possess a core conserved RCCCGC motif. In the 5′ UTR, one major subset of motifs consists of purine-rich repeats. A distinct and substantial fraction possesses upstream AUG start codons. Half of the AUG containing motifs reveal hidden protein-coding potential in the 5′ UTR, while the other half point to a peptide-independent function related to translation. Among the former, we added four novel peptides to the small catalog of conserved-peptide uORFs. Among the latter, our case studies document patterns of uORF evolution that include gain and loss of uORFs, switches in uORF reading frame, and switches in uORF length and position. In summary, nearly three hundred post-transcriptional elements show evidence of purifying selection across the eudicot branch of flowering plants, indicating a regulatory function spanning at least 70 million years. Some of these sequences have experimental precedent, but many are novel and encourage further exploration.
Keywords
INTRODUCTION
In most eukaryotic organisms tested, transcript concentration is only loosely correlated with protein concentration (Baerenfaller et al. 2008; de Sousa Abreu et al. 2009). In mammals and fungi, a substantial fraction of this decoupling results from gene-specific variations in translation efficiency (Ingolia et al. 2009; Vogel et al. 2010). The identification and categorization of elements responsible for this variation would help to assess their biological significance and to more fully understand the pathways in which they act. Thus, we employed a comparative transcriptomics pipeline in order to determine the prevalence and relative proportions of post-transcriptional regulatory sequences within flowering plants.
Upstream start codons (uAUGs) are among the most ubiquitous gene-specific elements affecting an mRNA's protein expression level (Calvo et al. 2009). Because ribosomes scan the mRNA in a 5′ to 3′ direction in search of a start codon, these uAUGs will, with variable frequency, become initiation sites for protein synthesis (Ingolia et al. 2009). Their associated open reading frame (uORF) may either overlap the major ORF (mORF) or terminate upstream of the mORF start codon (Fig. 1A). In either case, uORFs can drastically reduce protein expression (Calvo et al. 2009; Zhou et al. 2010). In some cases, uORFs are conserved at the peptide-level (Franceschetti et al. 2001; Hayden and Jorgensen 2007; Tran et al. 2008; Rahmani et al. 2009)—referred to as “conserved peptide uORFs” (CPuORFs). CPuORFs shared by Arabidopsis thaliana and rice fall into 19 homologous groups (Hayden and Jorgensen 2007). The degree to which uORFs are conserved over short or long evolutionary time scales is still not well-delineated, although uAUG triplets in mammals and fungi evolve slower than other 5′ UTR triplets (Churbanov et al. 2005; Neafsey and Galagan 2007).

(A) Cartoon of a generic plant mRNA with specific sequence regions and known post-transcriptional elements indicated. (B) Schematic of the computational pipeline used for conserved sequence identification and categorization. Tasks are described in each box. Outputs are unboxed and overlap an arrow if that output then becomes the input for a downstream processing step.
Many researchers have proposed that groups of specific genes are coregulated at the mRNA level by interactions between a common RNA binding factor and its cognate RNA sequence element (Gerber et al. 2004; Keene 2007). This “regulon” concept is supported by experiments on the PUF family of RNA binding proteins within complex eukaryotes (Gerber et al. 2006; Francischini and Quaggio 2009) and on 40 RNA-binding proteins in yeast (Hogan et al. 2008). A striking example is the PUMILIO protein (a PUF-family protein) in Drosophila, which interacts with mRNAs for a majority of subunits of the vacuolar ATPase (Gerber et al. 2006). The sequence specificity of PUF-family proteins appears to stay constant across major taxonomic divisions, while the function of their target genes can vary drastically (Gerber et al. 2006). Such variation appears to carry over into plants (Francischini and Quaggio 2009), but its full extent has yet to be determined.
The regulatory sequences described above reside in either the 5′ or 3′ UTR. Because of assorted codon usage constraints, it is difficult to assess peptide-independent nucleotide conservation within individual mORFs, particularly with regard to short motifs (Chen and Blanchette 2007). Therefore, we have focused on the UTRs. Respectively, repression by small RNAs (smRNAs) is another major regulatory process known to act post-transcriptionally on specific mRNAs, and, as opposed to metazoans, most putative smRNA target sites in A. thaliana are located in the mORF of mRNAs (Backman et al. 2008). Still, a few smRNA target sites do appear in UTRs, and these can be used to assess trends in target-site conservation without the confounding effects of conservation related to protein encoding.
Although post-transcriptional processes are sometimes mediated by RNA secondary structure, most mRNA-specific interactions require a primary nucleotide sequence component (Rabani et al. 2008; Serganov and Patel 2008; Gilligan et al. 2011). The conservation of such primary sequence elements is typically easier to interpret because similarity in secondary structure is often a result of nucleotide composition (Rivas and Eddy 2000) and, even when constrained structure can be identified via covariance, these structures are difficult to generalize across the genes that harbor them (McGuire and Galagan 2008; Rabani et al. 2008). Since most intergenic or untranslated DNA evolves at a neutral rate, primary sequences separated by a large evolutionary distance cannot be aligned with confidence. Additionally, at such distances, conserved motif identification is confounded by site turnover (Doniger and Fay 2007). The “window of useful divergence” to identify conserved noncoding sequences by alignment is ∼10–40 million years for plants (Freeling and Subramaniam 2009). To overcome these difficulties for the eudicot branch of flowering plants, which diverged as early as 70 million years ago, we assessed the statistical enrichment of motifs that are linked to orthologous coding sequences—a technique that is alignment-free and somewhat robust to site turnover or displacement (Blanchette and Tompa 2002). Our approach, based on the MEME algorithm (Bailey and Elkan 1994), is most effective when all sequences compared have undergone effectively randomizing divergence. Since we are looking for deep conservation, this prerequisite is satisfied by default (Galagan et al. 2005; Chen and Rajewsky 2006). Yet, when creating orthologous groups from distantly related plant species, in-paralogs resulting from local duplication or polyploidy can produce false positives caused by short divergence times (Blanchette and Tompa 2002). As described below, we solved this problem by reducing groups of orthologous sequences to combinatorial subgroups.
The identification of sequence conservation across orthologous intergenic or promoter regions is typically interpreted as functional constraint on transcription factor binding sites, while conservation of amino acids in a protein is interpreted as constraint relating to protein function. Because, as described above, variable forms of regulation can act via the UTR, it is easy to misinterpret conservation within the UTR. For example, smRNA binding sites can pose as CPuORFs if synonymous to nonsynonymous mutation bias is not assessed. Therefore, we have categorized conserved motifs into plausible functional groups based on whether or not they (1) have dinucleotide repeats that resemble microsatellites, (2) are target sites of known or predicted microRNAs, (3) code for a peptide that is evolutionarily constrained at the amino acid level, (4) qualify as putative recognition elements (PREs) based on pockets of dense conservation, and (5) match known transcription factor binding sites (Fig. 1B).
The motif identification approach was applied to six eudicot lineages, including tobacco, grapevine, soybean, orange, cotton, and Arabidopsis. These six species were chosen not only because each provides substantial transcript data but also because each marks a distinct, highly diverged, branch of the eudicots (Fig. 2A). Moreover, they represent many of the different life histories and characteristics of the eudicots: growth habit (tree, vine, perennial shrub, annual herbaceous plants), geographic origin and latitude, history of domestication, level of polyploidy, and genome size. We found that at least 3% of orthologous groups have one or more deeply conserved UTR motifs. It stands to reason that the majority of these motifs act at the post-transcriptional level. In spite of a similar nucleotide composition, the 5′ and 3′ UTRs have distinct complements of conserved motifs, as predicted by canonical models of eukaryotic translation. Elements that engage the ribosome are found preferentially in the 5′ UTR, while elements resembling protein binding sites are more prominent in the 3′ UTR. We find some evidence for the notion that groups of mRNAs form RNA regulons. However, such groups are typically from the same gene family. Our data suggest that many of the conserved sequence motifs regulate individual genes, rather than gene families, and many of the target genes are regulators of transcription, signaling, or protein turnover.
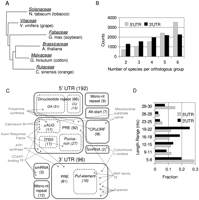
The landscape of post-transcriptional regulatory sequences that are conserved in flowering plants. (A) Tree representing descent and relative divergence of the species in this analysis. Modified from Bausher et al. (2006) and based on chloroplast genomes. The family name is given above its representative species. (B) Distributions for the number of species per orthologous group in 5′ and 3′ UTR comparisons. (C) A categorical map of conserved motif annotations. Box size roughly indicates the proportion of annotations that we assigned to each category. Exact values are given in parentheses. A box with dashed lines indicates that the category is a subset of the larger, solid-lined box. A dot indicates a single orthologous group. Only a small selection of orthologous groups are shown, and these are placed in the annotation box associated with their conserved motif. Gray labels describe the larger gene families to which these specific orthologous groups belong. A dashed line between orthologous groups indicates that the motifs are similiar (RAP family, Expansin). A dashed line added to Calcineurin B indicates multiple annotations of its conserved motif. (TFBS) transcription factor binding site. (D) Histograms, calculated independently, of PRE lengths in both UTRs. Related to Supplemental Files 2, 3, and 4.
RESULTS
Three percent of orthologous groups contain a conserved motif in the UTR
UTRs are difficult to predict computationally from genomic DNA (Brown et al. 2005); thus, experimentally confirmed mRNA sequences are needed for valid UTR comparisons. To this end, A. thaliana transcripts were acquired from TAIR (version 9). Putative transcript data for five informant species with >60,000 putative transcript entries were downloaded from PlantGDB (Fig. 2A; Supplemental Table S1). The PlantGDB transcripts were assembled from all available expressed sequence tag (EST) and cDNA sequence data in GenBank. The longest continuous reading frame containing an in-frame AUG for each transcript was considered the major ORF. mORFs were translated and clustered into orthologous groups using reciprocal blastp and OrthoMCL (Supplemental File 1). The sequence upstream of a mORF was considered its 5′ UTR. Likewise, the sequence downstream was considered its 3′ UTR.
Of the 11,887 groups containing an A. thaliana sequence, 10,122 (85.1%) contained at least one informant species as well (Supplemental Table S1). The remaining 14.9% of A. thaliana groups consist solely of in-paralogs, where no orthology across lineages could be inferred. This number, 10,122, is comparable to the number of orthologs, 10,381, shared by A. thaliana and Ricinus communis, both of which have sequenced genomes (Chen et al. 2006). This number is necessarily smaller than the total number of genes per genome because many plant genes are closely related in-paralogs derived from recent genome duplications. Therefore, the number of orthologous groups identified here approaches the theoretical maximum. For both UTR regions, the majority of comparisons have good representation, i.e., four to six species (Fig. 2B).
The 5′ and 3′ UTRs of an mRNA generally evolve faster than its main protein coding sequence (CDS). Given the divergence between the species in this study, neutrally evolving portions of the UTR will have a nominal number of consecutive bases conserved as a result of relatedness alone. We, therefore, used the MEME algorithm to search for enriched elements within these UTRs. MEME assumes a random background model and calculates an E-value based on the number of times a given motif is expected to be present by chance in a given set of sequences. Importantly, intra-species paralogs (in-paralogs), resulting from post-speciation duplication events, could potentially have undergone very short divergence times, undermining our assumption of effective randomization and disrupting the identification of conserved motifs. Also, many orthologous groups contain two or more alternative transcripts of the same gene, which are operationally indistinguishable from in-paralogs. To address these issues, all orthologous groups were subdivided combinatorially such that each comparison involved only one sequence from each species in the orthologous group (see Materials and Methods). This approach has two additional benefits: (1) In-paralogs, which may have undergone neo/subfunctionalization at the regulatory level by losing an element (Lockton and Gaut 2005), do not add noise to the identification process; and (2) our false discovery rate can be estimated by simply randomizing orthologous groups, as opposed to simulating mutations.
Our statistical criteria, defined in Materials and Methods, resulted in 194 and 96 conserved motifs for the 5′ and 3′ UTRs, respectively (Fig. 2C). Thus, 3% of orthologous groups have a deeply conserved primary sequence element. Based on randomization of orthologous groups, our false discovery rate was 6.1% (12/194) for the 5′ UTR and 3.1% (3/96) for the 3′ UTR (i.e., for the 3′ UTR, we expect ∼3 false positives per 100 positive results). We further characterized motifs identified by MEME based on their composition and their patterns of conservation.
[AG] and [CU] repeats are enriched in the 5′ UTR
Microsatellites (mono/dinucleotide repeats) are common in plants, have a regional bias in the genome (Lawson and Zhang 2006), and are conserved between orthologs and across paralogs (Zhang et al. 2006). The consensus sequence associated with each motif was checked for >5 consecutive mononucleotide repeats or >3 consecutive dinucleotide repeats. We found a dramatic enrichment of conserved dinucleotide repeats in the 5′ UTR relative to the 3′ UTR: 51-fold enrichment for [AG]n and 15-fold for [CU]n (Table 1). Mononucleotide repeats showed no such 5′/3′ bias. Randomization of orthologous groups (“random” data sets in Table 1) indicates that only a small proportion of conserved dinucleotide motifs can be attributed to the general enrichment of [AG] or [CU] repeats within 5′ UTRs. GA repeats are enriched around and downstream from the transcription start site in plant genes, and the motif is more often found in TATA-less genes, which have a broad cluster of transcription initiation sites (Yamamoto et al. 2009). Concerning the CU repeats, pyrimidine-rich motifs (Y-patches) are common in the 5′ UTR, but their function remains unclear (Molina and Grotewold 2005; Yamamoto et al. 2007, 2009).
Number of conserved repeat motifs in 5′ and 3′ UTRs
One-fourth of known A. thaliana smRNA target sites in the UTR are conserved, and their conservation profiles support the “seed” hypothesis
In striking contrast to metazoans, there are only 238 known small RNA target sites in the Arabidopsis small RNA project database (ASRP) (Fahlgren et al. 2007; Backman et al. 2008). Of these, only 15 target sites appear to be located in the 3′ UTR. Of the balance, eight occur in the 5′ UTR, while the remaining ones lie in the CDS of targeted mRNAs.
After removal of repeat motifs, we checked the A. thaliana representative of each of our motifs (with +/− 10 flanking nucleotides) against all putative smRNA target sites. Two of the eight known target sites in the 5′ UTR were conserved and seven of the 15 in the 3′ UTR (Table 2—only three entries are shown for the 3′ UTR because the remainder are found within the same orthologous groups). Motifs were also checked against more liberal predictions of smRNA targets and recently discovered miRNAs (Alves et al. 2009; Breakfield et al. 2011), with the same result. The “seed” hypothesis predicts that complementarity to the 5′ end of the mature smRNA is critical for mediating silencing (Brodersen and Voinnet 2009). To varying degrees, all five motifs support this hypothesis. All but three of the 15 genes with a known Arabidopsis 3′ UTR target site had the requisite sequence coverage for a valid comparison. In addition, all miRNA families listed in Table 2 are known to be conserved across their entire length in at least two dicots (Willmann and Poethig 2007). Therefore, the 3′ degeneracy of their target sites cannot be accounted for by covariation in the targeting smRNA. Together, these results suggest that many miRNA target sites appear to be lineage-specific.
Motifs implicated in experimentally confirmed smRNA-mediated degradation pathways
At least 18 conserved uORFs function at the peptide level
If any region in the UTR codes for a conserved protein, then that region will appear as a highly significant motif in our analysis. A conserved protein could take the form of a conserved-peptide uORF (CPuORF), a segment of main ORF downstream from a non-AUG start codon, or a segment of main ORF that was misannotated as 5′ UTR due to an ambiguous splicing pattern. We, therefore, checked all motifs for their coding potential. The longest continuous reading frame (CRF) for each of the three possible reading frames associated with a motif was aligned as protein. Each alignment was tested for coding potential based on the likelihood of nonsynonymous mutations being under negative selection (see Materials and Methods). After eliminating artifacts stemming from alternative splicing events, motifs were considered CPuORFs when significant protein-coding potential overlapped an explicit uORF (see Fig. 3).
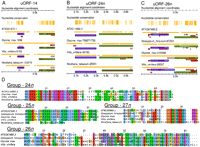
Novel CPuORFs. (A–C) Transcript alignments are shown in miniature followed by all possible ORFs, where the darkness of red indicates the context strength of the AUG. CPuORFs are bordered in blue; the beginning of each associated mORF is bordered in green. Orange vertical lines (“Nucleotide conservation” lane) indicate that all residues are identical in that column. Note gaps in sequence identity between uORF and mORF. The orange horizontal line below each accession indicates sequence coverage or indels relative to the alignment. In the scale bar, 0.5 k = 500 nt. Sequences are ordered, top to bottom, relative to their phylogenetic distance from A. thaliana. (A) Previously confirmed CPuORF-14 (Hayden and Jorgensen 2007). (B) CPuORF-24n. (C) CPuORF-26n. (D) Protein alignments of novel CPuORFs. Color scheme is ClustalX default. Within alignments, sequences are ordered relative to their distance from A. thaliana. Related to Table 3.
All motifs associated with protein-coding potential and their annotations based on the accession for the A. thaliana representative of the orthologous group
We identified four CPuORFs with little or no precedent in the literature—indicated by “n” in Table 3. These novel CPuORFs show similar conservation profiles and spatial patterns to known CPuORFs (Fig. 3A–C), although CPuORF-24n is particularly unusual in that it begins near the cap and extends the length of the entire 5′ UTR (Fig. 3B). CPuORFs 24n and 25n exhibit extensive amino acid conservation across their entire length, while the others show a 3′ bias in their degree of conservation (Fig. 3D). We also recapitulated 13 of the 19 CPuORFs found previously in an A. thaliana (dicot) and Oryza sativa (monocot) comparison (Table 3; Hayden and Jorgensen 2007). One might have expected to find a substantial number of dicot-specific CPuORFs, which would not have been detected in prior Arabidopsis-monocot comparisons. However, this was not the case. Among the four novel CPuORFs, three were found in one or more monocot lineages (Supplemental Table S2). Only one CPuORF, 27n, could not be found in any of three monocot lineages examined, even though extensive sequence data exists for the 5′ UTR of CIPK6 homologs. Therefore, it appears that CPuORFs are rarely lineage-specific with regard to the angiosperm phylogeny.
In addition to CPuORFs, seven protein-coding motifs were categorized as conserved “non-AUG starts” of the mORF, because they were consistently in-frame with the mORF and not explained as alternative/incomplete splicing artifacts (Table 3). The beginning of each of these putative coding regions contains a variant of the pattern “A(A|C)N(G|U|C)UGG,” where “N” indicates any nucleotide and brackets indicate possible residues (Supplemental Fig. S1). This pattern, excepting the variable adenine of the canonical translation start codon, matches the strongest Kozak context for translation initiation in plants (Lukaszewicz et al. 2000). In some prior cases, such sites are used to produce multiple protein isoforms for the same mRNA molecule, which can then be targeted to various organelles (Wamboldt et al. 2009). Also, for seemingly different reasons, a key regulator of flowering time in plants, FCA, consistently uses CUG as an alternative translation initiation site (Simpson et al. 2010). Our data confirm that non-AUG initiation requires a strong context. Moreover, they document that non-AUG initiation is not a fluke restricted to a specific lineage but, given its deep conservation, must have provided a selective advantage during much of the eudicot evolutionary history.
In the 3′ UTR, after comparable filtering criteria, we were left with five potential protein-coding motifs. None of the five were consistently frame-biased in terms of protein-coding potential, and so these likely do not represent conserved read-through or programmed frame-shift events. They may represent exons from rare isoforms or, given their short length (<10 codons), may have coding potential by chance. Given that plant viruses routinely make use of stop codon read-through to expand their protein-coding potential (Skuzeski et al. 1991; Urban et al. 1996), the absence of any evidence for conserved read-through events in these dicot lineages is noteworthy.
Putative recognition elements
Short sequences exhibiting strong conservation in MEME alignments can have equivalent or lower E-values than longer, weakly conserved, sequences. Though both may be important for plant function, one of our aims was to identify potential recognition elements, which experiments suggest are between five and 20 nt long (Farley et al. 2008; Hogan et al. 2008; Pagano et al. 2009). Motifs not belonging to any of the categories discussed above were scanned for regions of dense conservation (see Fig. 1). Within these motifs, the largest window of average consensus-letter frequency greater than 0.92 (see Materials and Methods) was defined as a putative recognition element. See Supplemental File 2 for a complete list and Supplemental Files 3 and 4 for local MEME alignments.
We identified 92 PREs in the 5′ UTR and 81 in the 3′ UTR (Fig. 2C). Motifs in the 5′ UTR are generally shorter than motifs in the 3′ UTR (Fig. 2D). In the 3′ UTR, PREs peak at ∼20 nt in length. Again, these conserved motifs have already been filtered to remove likely smRNA target sites; hence, though this is the size expected for smRNA target site conservation (as in Table 2), we consider these to represent non-smRNA binding elements. In both regions, PREs make up the majority of conserved motifs (Fig. 2C). In a gene ontology (GO) term analysis, genes with a 3′ UTR PRE are significantly overrepresented in two specific categories: proteins targeted to cell periphery (20 genes, 3.0-fold enrichment, p-value = 4.44 × 10−3) and signal transduction proteins (12 genes, 4.2-fold enrichment, p-value = 1.80 × 10−2). Genes with 5′ UTR PREs are not distinctly enriched in any category.
PUF-binding motifs, possible Expansin localization signals, and other novel elements are conserved in the 3′ UTR
PREs are derived from conserved motifs associated with an individual orthologous group. Yet, it is likely that certain PRE patterns are shared between multiple orthologous groups. To generalize PREs based on their shared sequence content, we assessed the degree to which 4-mer words were enriched among all PREs. We created random expectation distributions (n = 10,000) for each 4-mer such that the length distribution of the PRE data sets was recapitulated. We then assessed the p-value of the actual enrichment based on its position within this distribution. The 3′ UTR and 5′ UTR regions were treated independently.
Nearly all PUF-binding elements in metazoans, plants, and yeast contain a core UGUA sequence (Gerber et al. 2004, 2006; Francischini and Quaggio 2009). Indeed, UGUA was one of the most significantly enriched 4-mers in the PREs of the 3′ UTR and, notably, not in PREs of the 5′ UTR (Table 4). Sixteen of the ∼80 genes have UGUA-containing PREs in the 3′ UTR (Table 5). These are most likely authentic PUF binding sites for the following two reasons. First, one of these, LHCa4, was one of five mRNAs found to interact with the PUF protein, APUM2, in a yeast three-hybrid screen (Francischini and Quaggio 2009). Another protein identified in the same screen, a DNAJ, also has a conserved UGUA motif but with an E-value (6.7 × 10−5) that did not pass our conservative statistical cutoff. Of the remaining three APUM2 targets, all have orthologs in at least four species with 3′ UTR coverage, but the UGUA element is not conserved. Second, the mRNA of CLAVATA1 (At1g75820), which is critical for meristem maintenance, was also shown to interact with APUM2 (Francischini and Quaggio 2009). Indeed, the CLAVATA1 3′ UTR contains four UGUA tetramers, although each individual motif had a high E-value in our analysis (data not shown). However, At4g20270.1 is BAM3, an in-paralog of CLAVATA1, and its longer UGUA-PRE is highly conserved (Table 5). These results show that our pipeline was able to identify a subset of authentic 3′ PREs that bind PUF-domain RNA binding proteins. Generally, the UGUA-containing PREs appear to be dispersed among functionally unrelated mRNAs, except for the two subunits of the photosystem I light harvesting complex, AT3G47470.1 and AT3G61470.1 (Table 5). Notably, transcripts associated with photosynthesis are known to be enriched around the chloroplast (Marrison et al. 1996), although the mechanism is still unknown.
4-mer words significantly enriched in PREs from 5′ and 3′ UTRs
Possible PUF-binding PREs within the 3′ UTR
The UUUG 4-mer is enriched in 3′ UTRs to a similar extent as UGUA (Table 4). These two 4-mers are occasionally found together but not in a consistent arrangement relative to one another (Table 5). They may be functionally independent. Moreover, their co-occurrence does not deviate from what would be expected given their individual distributions among all PREs (p-value = 0.30, χ2 test), suggesting that UUUG is generally unrelated to PUF-binding.
Expansins are one of the few groups of plant mRNAs that have been shown to be localized to specific subcellular sites (Im et al. 2000). A conserved [AG]CCCGC-containing motif was found in four out of 12 available Expansin 3′ UTRs (Fig. 4A). A region upstream of the [AG]CCCGC motif is also conserved but more specific to each Expansin. The Zinnia elegans Exp1 mRNA (gi|7025490|gb|AF230331.1), which was shown to localize to a particular region of the cell periphery (Im et al. 2000), also contains this pair of elements (Fig. 4B). They, too, appear to be position-independent relative to the mORF stop site and relative to one another. Taken together, Expansin mRNAs may form a small RNA regulon, where a common type of motif drives the coregulation of several distinct mRNAs, but where a secondary motif confers some degree of additional specificity.
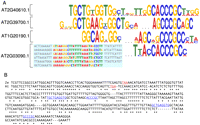
Expansin 3′ UTRs contain a combination of conserved sequences, which are also present in the 3′ UTR of the localized Zinnia elegans Exp1 mRNA. (A) Sequence LOGO plots are generated by MEME; information content of a position is represented by stack height, which is multiplied by letter frequency at that position to give letter height. Each Expansin mRNA has one upstream variable region and one downstream RCCCGC-core motif. The entire MEME-derived alignment is given for AT2G03090.1 group's variable region. (B) Alignment of the Zinnia elegans ExpansinA1 mRNA (Ze)—gi|7025490|gb|AF230331.1—which is targeted to specific subcellular sites, with its A. thaliana ortholog (At)—AT2G40610.1. mORF stop codons are in red, and conserved elements are in blue. Asterisks indicate that the column letters are identical. Note: (U)racils are shown as (T)hymines.
5′ UTR PREs are enriched in purine-rich 4-mers
In an attempt to better classify and characterize the 5′ PREs, we examined which 4-mer words were overrepresented. The most significant 4-mers among the 5′ UTR PREs are purine-rich words (e.g., AGAA) (Table 4). These purine-rich PREs are in a continuum with the [AG] repeat motifs that were set aside early in the motif extraction pipeline. We explored whether the motifs identified here function at the transcriptional or post-transcriptional level. First, [AG] repeats can act at the transcriptional level (Santi et al. 2003; Meister et al. 2004; Kooiker et al. 2005). Purine-rich motifs are also present in many 5′ UTRs and are associated with broad clusters of transcription start sites (Yamamoto et al. 2009). However, purine-rich repeats in the 5′ UTR have also been reported to enhance translation of the ntp303 gene in N. tabacum (Hulzink et al. 2002). Using mRNA half-life data from a prior study (Narsai et al. 2007), we found that transcripts containing a purine-rich PRE in the 5′ UTR have a median half-life of 9.6 h, which is more than twice as long as the median half-life of 3.8 h for Arabidopsis transcripts in general (p < 10−5, sign test). No other PRE category correlated with RNA stability. This result is consistent with a new role of the purine-rich PRE in RNA stability.
To a lesser extent, pyrimidine-rich words—UUCU, UUUU, UCUU—are more common than expected in the 5′ UTR (Table 4). A few of these PREs resemble “Y-Patch” promoter elements, which can extend into the 5′ UTR (Yamamoto et al. 2007), although it is still unclear if Y-patches are acting post-transcriptionally or transcriptionally. In metazoans, the expression of ribosomal protein mRNAs is governed by 5′ terminal oligopyrimidine motifs (Avni et al. 1994), and translation of mRNAs with this feature is favored in wheat cell lysates (Shama and Meyuhas 1996). Yet, we find no ribosomal proteins with conserved CU-rich elements (Supplemental Files 2 and 3).
We examined whether 5′ PREs are likely to represent transcription factor binding sites. The Telobox (AAACCCUA or its reverse complement) was found in six of the 5′ PREs (Supplemental File 2). Because this motif occurs commonly in both orientations around plant transcription start sites (Tremousaygue et al. 2003; Molina and Grotewold 2005), it is a candidate for functioning at the DNA level.
Based on the AGRIS database of A. thaliana promoter motifs (Davuluri et al. 2003), only 11 out of 92 5′ UTR PREs have evidence for containing transcription factor binding sites (Supplemental File 2; Fig. 2C). For comparison, 3′ UTRs contain few confirmed promoter motifs, yet eight out of 96 PREs would be annotated as transcription factor binding sites (Supplemental File 2). A similar result was obtained from the AthaMap project (Bülow et al. 2010) (data not shown). Careful inspection of the 11 cases revealed that the most conserved core of the PRE either lies beside the transcription factor motif or extends beyond the motif (data not shown). Therefore, it seems likely that few of our 5′ UTR PREs are acting as transcription factor binding sites.
Conservation and variation of 5′ PREs reveal patterns of uORF evolution
The 4-mer associated with a strong-context start codon, AUGG, is significantly enriched among PREs in the 5′ UTR (Table 4). In fact, many AUGs appear within conserved motifs, in both weak and strong contexts (Table 6). Since sequences with extensive conserved protein-coding potential were already removed prior to this portion of the analysis, these 4-mers are conserved, at least in part, for peptide-independent effects relating to uAUG initiation/reinitiation.
uAUG-containing PREs within the 5′ UTR
Because the effect of uAUGs on gene expression is undoubtedly via translation, we focused on AUG-containing 5′ PREs to glean a few trends governing the evolutionary changes in 5′ PREs and their associated uORFs (Fig. 5; Supplemental Fig. S2). As the first trend, when a uORF overlaps the major ORF, their relative frame arrangement is usually conserved (AS1, PRR2/TOC2, smRNP At3g14080). In contrast, uORF clusters that do not overlap the mORF lack a consistent frame arrangement with the mORF (e.g., S6K, CBL9, NF-YA10, HD-ZIP-6, At1g19330, ADC). This pattern is quite striking in the case of smRNP, where all three overlap-uORFs occupy the −1 frame with respect to the mORF. In contrast, four nonoverlap uORFs occupy two different frames.

Conservation and variation in the 5′ UTR of mRNAs that harbor uAUG containing PREs and/or uORFs. Each subpanel shows the uORF pattern (colored rectangles) and, where present, the PRE (tall blue box) of one orthologous group; some orthologous groups were supplemented manually with members from additional species. The Gene Identifiers come from TAIR (At#g#####), PlantGDB (PUT…), or GenBank (all others). The start of the major ORF is symbolized by a white box. The 5′ upstream region is indicated by a thick line. Vertical lines inside the uORF stand for internal in-frame AUG codons. Unless indicated, identical color or number does not imply sequence similarity. uORFs centered on the UTR are in the same frame as the major ORF, while those above the line and below the line are in the −1 and +1 frames, respectively. Selected exon-exon junctions are indicated by black arrowheads. Species abbreviations are as follows: (At) Arabidopsis thaliana, (Cs) Citrus sinensis, (Gh) Gossypium hirsutum, (Gm) Glycine max, (Nt) Nicotiana tabacum, (Vv) Vitis vinifera. (A) Calcineurin CBL3 and CBL2. This 5′ UTR contains a highly conserved PRE with two AUGs in a conserved frame arrangement. The downstream uAUG leads into a uORF that overlaps the main ORF (orange). A third uORF is present in some species (yellow). Its uAUG overlaps the stop of uORF1. (B) ASSYMMETRIC1/PHANTASTICA. This 5′ UTR lacks a recognizable PRE, yet its uORF pattern is highly conserved. Invariable features include one overlap uORF (blue) and one upstream uORF (yellow). An additional uORF ends at the start codon of the main ORF (green); it is present in all species except soybean. The consistent pattern notwithstanding, the uORF nucleotide and peptide sequences are not highly conserved. (A. majus) Antirrhinum majus, (A. lyrata) Arabidopsis lyrata. (C) PSEUDO-RESPONSE REGULATOR2/TOC2. This 5′ UTR harbors a conserved PRE with a conserved uAUG that leads into a long overlap-uORF (green). An additional uORF is usually present upstream, except in Arabidopsis. (D) RING protein At1g71980. Its PRE invariably harbors the start codon of a short uORF (green). A second uAUG is usually present in the PRE (orange), except in Arabidopsis. Additional short uORFs are common. (E) smRNP protein At3g14080. A PRE with a conserved uAUG leads into a uORF of variable length (green), which may overlap the major ORF. Upstream uORFs may be present. In Arabidopsis, the uAUG and uORF have been lost from the PRE. (F) Spermidine synthase. The PRE contains a putative non-AUG start codon. The non-AUG uORF downstream (orange, stippled) is highly conserved in length and amino acid sequence (data not shown). Related to Supplemental Figure S2.
Second, in cases where the PRE harbors multiple AUGs, their relative reading frame arrangement is generally highly conserved. Examples include calcineurin CBL3, myb protein AS1 uORFs 2 and 3, RING At1g71980, CCAAT enhancer binding factor NF-YA10, and expressed protein At1g19330. In contrast, uORFs that are not initiated in the PRE tend to have a variable frame position with respect to the PRE's uORFs (PRR2 uORF2, RING At1g71980 uORF3). Although we selected for situations like this by focusing on conserved PREs, the trend is, nevertheless, noteworthy. Because the initiation at a given AUG is strongly affected by an overlapping uORF (Hanfrey et al. 2005; Roy et al. 2010), we suspect that most of these uORFs modulate the strength of initiation among each other and at the major ORF.
Third, in the case of PREs that do not specify overlap-uORFs, the relative position of the PRE in the 5′ UTR appears to be somewhat restrained (RING, CBL9, HD-ZIP-6, At1g19330, ADC), even though the remaining 5′ UTR sequences have usually diverged to the point that they cannot be aligned.
Fourth, the uORF pattern in Arabidopsis and other Brassicaceae often differs from the consensus in the other families. Examples can be seen in S6K, PRR2/TOC2, the smRNP, the RING At1g71980, NF-YA10, and HD-ZIP-6.
Fifth, in-paralogs often differ in their PRE and uORF pattern. Examples include Arabidopsis S6K, calcineurins CBL9 and CBL1, NF-YA10 and 2, HD-ZIP-6 and 16, expressed protein At1g19330, ADC, and CTR1.
Together, these case studies show that uORFs associated with PREs tend to be highly conserved, whereas the uORFs that reside outside of PREs tend to be quite variable. However, even uORFs associated with PREs have been subject to significant variation during evolution of the Brassicaceae, and even otherwise highly conserved uORFs tend to vary between in-paralogs. All of these observations suggest that uORFs are one of the dials of molecular evolution that alter gene expression levels such that they are fine-tuned to be adaptive in specific lineages.
DISCUSSION
Patterns of element enrichment in the 5′ versus 3′ UTR reflect the canonical model of eukaryotic translation
Judging by the distribution of PREs, the 5′ and 3′ UTRs mediate distinct forms of post-transcriptional regulation. This is expected given the distinct molecular events occurring within each region during translation (Jackson et al. 2010). Scanning of the small ribosomal subunit through the 5′ UTR is likely to displace transient interaction between RNA and trans-acting factors whereas no such restriction applies to the 3′ UTR, which is thought to be free of ribosome traffic (Gu et al. 2009). The 5′ UTRs retain sequences that engage the ribosome itself, i.e., CPuORFs, non-CPuORFs and non-AUG initiation sites. The 5′ UTRs also harbor purine-rich and pyrimidine-rich repeat sequences (Fig. 2C). The remaining PREs in the 5′ UTR tend to be short (Fig. 2D). In contrast, the 3′ UTR largely lacked conserved peptides and AUG-related motifs, and low-complexity repeat motifs were uncommon. Instead, the 3′ UTR is where more complex PREs are found (Fig. 2C). Among these, putative binding sites for PUMILIO-type RNA binding proteins were detected only in the 3′ UTR. In the case of Expansin mRNAs, which must harbor a determinant responsible for their subcellular targeting, a candidate PRE was discovered in the 3′ UTR (Fig. 4).
Conserved target sites for smRNA are an exception to this trend of regional bias (Table 2). This is in striking contrast to metazoans, where 3′ UTR sites are much more common. Experiments on select genes in A. thaliana indicate that the position of a smRNA target site—3′ UTR vs. 5′ UTR vs. CDS—does not correlate with the degree to which an affected mRNA is degraded and/or translationally silenced (Brodersen et al. 2008), so perhaps a lack of regional bias is to be expected.
One extension of the RNA regulon concept is that, analogous to transcriptional modules, some mRNAs will be under combinatorial control by a suite of factors. Overall, only 8% (n = 7) of 5′ UTRs harbor legitimate examples of multiple conserved motifs (Supplemental File 2). In contrast, 16% (n = 13) of PRE-containing 3′ UTRs have significant conservation outside of the most conserved PRE, and generally this conservation is longer and more informative (Supplemental File 2). This pattern supports a model in which the 3′ UTR, because it is free of ribosomes, is a more appropriate platform for combinatorial regulation and signal integration than the 5′ UTR.
Identification of novel CPuORFs
We identified four novel CPuORFs, at least three of which are conserved in dicots and monocots, i.e., pan-angiosperm. The extensive length and conservation of the newly identified CPuORFs 26n and 24n (Fig. 3B–D) begs the question of whether CPuORFs are, in fact, regulatory elements or, alternatively, whether their associated mRNAs should be considered bicistronic transcripts. Strikingly, CPuORF-24n is identical to GenBank accession CDC26, a small subunit of the anaphase promoting complex (APC) (Dong et al. 2007; de F Lima et al. 2010). CPuORF-24n/CDC26 appears as a distinct transcript in metazoans and fungi, but in plants it is consistently found as a uORF upstream of a mORF that is annotated as adenylate cyclase. Notwithstanding that the existence of adenylate cyclase and cyclic-AMP in plants is controversial, as it stands, the associated mRNA is a good candidate for a multicistronic eukaryotic transcript. Yet, the conserved synteny, proximity, and cotranscription of CPuORF-24n/CDC26 and the putative adenylate cyclase suggest that, in plants, these two proteins require cotranslation.
Of the remaining three CPuORF loci identified in this study, little is known. CPuORF-27n is linked to a protein kinase involved in salt stress tolerance, and CPuORF-26n is upstream of an uncharacterized F-box protein. CPuORF-25n is associated with a dicarboxylic acid transporter in mitochondria.
What do conserved RNA sequence motifs reveal about the architecture of post-transcriptional regulation?
Certain genes and gene families are tightly associated with specific types of RNA sequence elements; the CPuORFs in the bZip transcription factor family are well-known. The entire polyamine synthesis pathway is a “hotspot” for translational control that incorporates CPuORFs, non-CPuORFs, non-AUG uORFs, and ribosomal frameshifting (Ivanov et al. 2010). In plants, thermospermine synthase is regulated by a uORF-dependent transcription factor (Imai et al. 2006), and arginine decarboxylase contains a conserved uORF (Chang et al. 2000), as do several catabolic polyamine oxidases (Hayden and Jorgensen 2007; Table 3; Supplemental Fig. S2). Aside from the well-investigated CPuORF in S-adenosylmethionine decarboxylase (Table 3) that reduces translation through polyamine-induced ribosome stalling, we have identified a PRE in arginine decarboxylase, a rate-limiting enzyme in the plant polyamine synthesis pathway (Supplemental Fig. S2), and a PRE in plant spermidine synthase (AT5G53120) (Supplemental File 3; Fig. 5F). Interestingly, this PRE contains the best match of all 5′ PREs to the noncanonical start site, AgaCUGG, possibly representing a uORF with a non-AUG start codon.
A second example for a focal point of RNA-level control that arose from this study is the CCAAT enhancer binding proteins. Two CCAAT-binding transcription factors have conserved microRNA target sites in the 3′ UTR (Table 2). Additionally, two other family members have PREs located in the 5′ UTR (Table 6; Supplemental File 2 and 3). Of these, the PRE of At3g05690 may possess a peptide-independent uORF. Notably, mammalian CCAAT-binding transcription factors also possess uORFs, which, in this case, govern CDS start-site selection (Calkhoven et al. 1994; Lincoln et al. 1998). This finding suggests that CCAAT-binding protein joins a small number of other genes, including S-adenosylmethionine decarboxylase and bZip transcription factors, which are regulated by uORFs across the plant-metazoan divide.
Also of note are the calcineurins. The subtype comprising Arabidopsis CBL1 and CBL9 has a 5′ PRE with a variable uORF, and the PRE in CBL9 can be recognized by the seed region of miRNA847 (data not shown); whereas the subtype comprising CBL2 and CBL3 has a 5′ PRE with a highly conserved uORF cluster. Finally, among the cytochrome c oxidases two family members have seemingly different modes of post-transcriptional control; AT3G15640.1 has a conserved microRNA binding site in the 5′ UTR (Table 2), while AT2G07687.1 has an extensive but clearly unrelated PRE in the 3′ UTR (Table 5). Such elements could be the basis for differential regulation.
Concerning the concept of RNA regulons, one expectation is that multiple, functionally related mRNAs share the same type of PRE. Again, the common sucrose-regulated CPuORF found in several bZip transcription factor mRNAs is a valid example. Here, we took note of a common PRE found in four Expansin mRNAs, including Expansins known to be targeted to specific subcellular sites.
It has been previously reported that RNA-binding proteins, among them multiple PUF-family proteins, associate with functionally related sets of genes (Hogan et al. 2008). Indeed, in our analysis, we find that LHCa2 and LHCa4, closely related genes, both contain a UGUA-PRE, a putative PUF-binding site.
Overall, however, we find very few instances of conserved coregulation of functionally related genes by the same motif and no examples to match the coregulation of the vacuolar ATPase subunits in Drosophila (Gerber et al. 2006). For example, genes with conserved UGUA-containing PREs were not significantly enriched in any GO category (data not shown). Undoubtedly, we have false negatives resulting from a lack of sequence coverage in the 3′ UTR, but genome comparisons within the fungal genus Aspergillus revealed only 48 such conserved PUF-binding sites, and the associated genes also do not have a GO bias (Galagan et al. 2005). This situation begs the question whether binding sites discovered via functional assays show evidence for selection in comparative genome sequence analyses. Many legitimate, sequence-specific mRNA-protein interactions may have little bearing on the fitness of the organism in which they occur. Differentiating those interactions that are neutral happenstance from those that are not will be critical in understanding not just how post-transcriptional control works but why it is used at all. Additionally, novel UTR sequences are fast emerging as a byproduct of extensive plant transcriptome sequencing. We anticipate that the techniques and motifs described herein will serve as an entrée into those data.
MATERIALS AND METHODS
Sequence acquisition and preparation
See Figure 1 for a general guide to the following computation pipeline. Transcript data for A. thaliana were downloaded from http://www.arabidopsis.org/help/helppages/BLAST_help.jsp#datasets on 24 September 2009 (Version 9). Putative transcripts for all other plant species were downloaded from http://www.plantgdb.org/prj/ESTCluster/progress.php on 7 October 2009 (Duvick et al. 2008). The PlantGDB versions of each species are as follows: Gossypium hirsutum, PUT-165a; Citrus sinensis, PUT-167a; Glycine max, PUT-169a; Vitis vinifera, PUT-173a; Nicotiana tabacum, PUT-169a. None of sequence sets have undergone significant additions. The longest ORF from each putative transcript was extracted using a custom Perl module (uORF.pm; all scripts are available at http://web.utk.edu/∼jvaughn7/code/) based on the criteria that an AUG be followed in-frame by a UAA, UGA, or UAG or that an ORF extend to the end of the putative transcript. These ORFs were translated to peptide sequences and reciprocal BLAST searched, using blastp (version 2.2.16), in species-wise fashion with an E-value cutoff of <10−30. In order to diminish confounding effects of lineage specific gene loss or incomplete sequence data on orthology assessment, accessions were then clustered based on their BLAST scores using OrthoMCL (version 1. 4) (Li et al. 2003), with default parameters. 5′ or 3′ UTRs shorter than 8 nt were excluded from analysis. Sequences completely overlapped by a larger, identical sequence were subsumed into that sequence.
Motif identification
We used MEME (version 4.3.0) (Bailey and Elkan 1994) to search for overrepresented sequences across the UTRs associated with orthologous groups of coding sequences (Wels et al. 2006). Because in-paralogs/alternative-transcripts within a group confound direct interpretation of E-values produced by MEME, each orthologous group was divided into all possible subgroups, where each subgroup contains only one sequence per species. For tractability reasons, if there were more than 30 possible subgroups per orthologous groups, only 30 randomly selected subgroups were processed further. For example, if an orthologous group contained four A. thaliana, three G. max, and six V. vinifera sequences, there would be 4 × 3 × 6, or 72 subgroup combinations; only 30 of these would be randomly selected for further evaluation. Only 11% of orthologous groups were large enough to require such reduction. Again, because of incomplete sequence information or lineage specific loss, we used the MEME option that detects zero or one motif in all sequences (“zoops”), where motifs could be 6–30 nt long. A second-order Markov model based on all A. thaliana 5′ UTRs (or 3′ UTRs, depending on the region being searched) was used as a background model in all MEME searches (Fan et al. 2009). To correct for multiple tests, we divided an E-value cutoff of 0.05 by the number of orthologous subgroups compared: 91,331 for 5′ UTR and 73,893 for 3′ UTR. Only the lowest scoring subgroup of an orthologous group was processed further. A motif was excluded from further analysis if the A. thaliana representative had a p-value of >10−9 of belonging to the motif by chance. Also, motifs were excluded from 5′ UTR comparisons unless the mORF proteins for >70% of informant species aligned, using ClustalW with default parameters, to within 25 amino-acids of the 5′ terminus of the A. thaliana representative. All analyses were also carried out on 5′ and 3′ UTR control data sets, which were generated by randomizing all orthologous groups such that species composition per orthologous group was maintained: all sequences were pooled based on the species to which they belong, and, for every orthologous group, the actual sequence was replaced with another sequence, drawn at random with replacement, from the same species pool. These data sets were used to determine our false discovery rate.
Though not reported in detail, we tested numerous alternative motif-identification algorithms. Generally word-based approaches such as Weeder (Pavesi et al. 2004) and FootPrinter (Blanchette and Tompa 2003) were less sensitive than MEME, presumably because they are less tolerant of site degeneracy. For example, at a comparable false discovery rate, Weeder found 12 enriched motifs in the 5′ UTR compared to 194 by MEME. Additionally, algorithms that took phylogeny into account, PhyloGibbs (Siddharthan et al. 2005) and PhyloCon (Wang and Stormo 2003), had equivalent or lower sensitivity than MEME, which is expected given that the lineages under analysis are highly diverged (Storms et al. 2010).
Motif categorization
The sequence of the most frequent letter at each site (consensus sequence) associated with each motif was checked for >5 consecutive mononucleotide repeats or >3 consecutive dinucleotide repeats (Supplemental Table S3). These motifs were removed from downstream processing. Next, the A. thaliana representative with +/−10 flanking nucleotides from each significant motif was searched against known and predicted smRNA target sites using the Arabidopsis Small RNA Project (ASRP) database (2 June 2010) (Backman et al. 2008) and data from Alves et al. (2009).
All possible open reading frames associated with a motif were checked for protein-coding potential. Each continuous reading frame from the A. thaliana representative was translated to protein and aligned using pairwise BLAST (b2seq) to every other sequence (also translated) of the appropriate frame in the MEME alignment. Only b2seq alignments with an E-value of <0.01 were considered homologous. All sequences passing this criteria were then aligned together using ClustalW (version 1.82), back-translated, and assessed for purifying selection against nonsynonymous mutations using PAML (version 3.14) (Yang 1997) according to the protocol in Nekrutenko et al. (2002) and a tree topology based on Figure 2A (Bausher et al. 2006). In brief, a likelihood ratio test was evaluated for a model in which the nonsynonymous (dN) to synonymous substitution (dS) ratio was allowed to vary and another model in which it was fixed at 1. Motifs with resulting p-values, from the χ2 distribution, of <0.01 were considered protein-coding motifs. Additionally, the motif was removed as an artifact of alternative splicing if the A. thaliana peptide associated with the coding potential had a blastp match (E-value cutoff of 10−5) to any Viridiplantae protein in the GenBank Refseq database that was 1.5 times longer than itself. The same analysis was done with explicit uORFs that overlap the motif. These were considered CPuORFs if, by manual curation (as in Fig. 3), their nucleotide conservation was exclusively associated with the uORF.
To identify PREs, we further differentiated remaining motifs based on the largest window in a positional weight matrix for which the average of all highest scoring letters for each column in the window was >0.92 (searched from left to right). The positional weight matrix was supplied by MEME; each letter in the matrix represents the frequency of that letter in the site. Windows that were >4 nt long, that were not known smRNA targets, did not show coding potential, and did not contain mono- or dinucleotide repeats were annotated as PREs.
All possible 4-mer word frequencies were measured separately for the combined sets of 5′ and 3′ PREs. The most frequent word was removed, leaving a gap to prevent artifactual fusions, and then the search was rerun until no more 4-mers existed. This prevents confounding effects of 4-mer overlap—UGGA overlapping GGAA, for example. Null distributions for each 4-mer were then generated by (1) randomly simulating a sequence data set with per-element length intact, (2) assessing the frequency of 4-mers, and (3) repeating the process 10,000 times. Because our initial inference of conservation accounts for higher-order statistical properties of the UTR, each letter in these randomizations was considered equally probable. An actual 4-mer frequency was placed within its null distribution, and the number of null distribution values greater than the actual 4-mer frequency was divided by 10,000 to get the p-value.
To further differentiate between PREs acting at the transcriptional versus post-transcriptional level, we searched the AGRIS database (Davuluri et al. 2003) in the manner described above for smRNA-related motifs. The IUPAC form used for AGRIS elements was converted to a regular expression prior to searching. Any A. thaliana regions associated with a PRE, as with the smRNA search above, were also checked again for imperfect matches to mature smRNAs using psRNATarget web server (Dai et al. 2010) with default parameters. Significant matching smRNA families are reported in Tables 5 and 6.
GO term enrichment
GO term enrichment was performed using the Amigo term enrichment web service with default parameters and all A. thaliana genes with a GO annotation as the null model (“TAIR” background filter). All A. thaliana accessions associated with the 5′ UTR PREs were pooled and searched; likewise for 3′ UTR PREs. Only one member per orthologous group was included.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the US Department of Energy through grant DE-FG02-96ER20223 (most figures), as well as the NSF Arabidopsis 2010 project DBI-0820047 (Table 2; Fig. 5; Supplemental Fig. S2). We thank Timothy Bailey, Charles Grant, and Jon Duvick for generous advice.
Footnotes
-
↵4 Corresponding author.
E-mail vonarnim{at}utk.edu.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031179.111.
- Received October 31, 2011.
- Accepted November 21, 2011.
- Copyright © 2012 RNA Society








