
Genome-wide evolutionary analysis of the noncoding RNA genes and noncoding DNA of Paramecium tetraurelia
- 1Institut de Biologie Animale Intégrative et Cellulaire, Université Paris Sud, Orsay 91405, France
- 2Centre National de la Recherche Scientifique (CNRS), UMR 8080, Orsay 91405, France
- 3Key Laboratory of Gene Engineering of the Ministry of Education, State Key Laboratory for Biocontrol, Zhongshan University, Guangzhou, 510275, People's Republic of China
Abstract
The compact genome of the unicellular eukaryote Paramecium tetraurelia contains noncoding DNA (ncDNA) distributed into >39,000 intergenic sequences and >90,000 introns of 390 base pairs (bp) and 25 bp on average, respectively. Here we analyzed the molecular features of the ncRNA genes, introns, and intergenic sequences of this genome. We mainly used computational programs and comparative genomics possible because the P. tetraurelia genome had formed throughout whole-genome duplications (WGDs). We characterized 417 5S rRNA, snRNA, snoRNA, SRP RNA, and tRNA putative genes, 415 of which map within intergenic sequences, and two, within introns. The evolution of these ncRNA genes appears to have mainly involved purifying selection and gene deletion. We then compared the introns that interrupt the protein-coding gene duplicates arisen from the recent WGD and identified a population of a few thousands of introns having evolved under most stringent constraints (>95% of identity). We also showed that low nucleotide substitution levels characterize the 50 and 80–115 base pairs flanking, respectively, the stop and start codons of the protein-coding genes. Lower substitution levels mark the base pairs flanking the highly transcribed genes, or the start codons of the genes of the sets with a high number of WGD-related sequences. Finally, adjacent to protein-coding genes, we characterized 32 DNA motifs able to encode stable and evolutionary conserved RNA secondary structures and defining putative expression controlling elements. Fourteen DNA motifs with similar properties map distant from protein-coding genes and may encode regulatory ncRNAs.
Keywords
- ciliates
- evolutionary constraints
- short introns
- noncoding RNA (ncRNA) genes
- untranslated regions (UTRs)
INTRODUCTION
Noncoding DNA (ncDNA) is functionally important in many respects. ncDNA harbors hundreds of noncoding RNA (ncRNA) genes in each genome, the transcription of which provides transcriptomes with functional ncRNAs. Pioneering studies identified the ribosomal RNAs (rRNA), small nuclear RNAs (snRNAs), and transfer RNAs (tRNAs) that are involved in messenger RNA (mRNA) splicing and translation, but ncRNAs are now also known to be involved in regulations as varied as gene transcription, chromosome replication, RNA processing, RNA modification, mRNA stability, protein degradation, and protein translocation (Storz 2002; Hannon et al. 2006). In addition, ncDNA harbors elements regulating gene expression and chromosome structure.
Computational efforts during the recent years have been directed toward the discovery of ncDNA elements and functional ncRNA genes. With the advent of genome-wide sequences, comparative genomic approaches have been developed. In mammals, these approaches led to the identification of segments >200 base pairs (bp) that are absolutely conserved between the human, mouse, and rat genomes. More than 100 of these segments, also known as ultraconserved elements, are located nearby genes or within introns (Bejerano et al. 2004). Recent studies have suggested that ultraconserved elements play a functional role in vertebrate genomes (Katzman et al. 2007), although some ultraconserved elements do not apparently provide any function (Ahituv et al. 2007).
Whole-genome duplications (WGDs) are now known to be common events of the eukaryote genome evolution. Subsequently to WGDs, single-copy loci arise from the sorting-out processes that reduced the genomes, while new functions are provided by the evolution of some protein-coding gene duplicates (Steinke et al. 2006). In the yeast Saccharomyces cerevisiae or in the fishes Tetraodon nigroviridis or Takifugu rubripes, 12%–15% of the protein-coding genes evolved from duplicates formed within a WGD (Jaillon et al. 2004; Kellis et al. 2004; Brunet et al. 2006). In the Arabidopsis thaliana genome that arose throughout three WGDs, the fraction of the protein-coding genes that arose from the last WGD and that are still in pairs is also 12%–15% (Vision et al. 2000; Simillion et al. 2002). Comparative analysis of ohnologs (duplicates resulting from WGDs) could be expected to be of great benefit in identifying and characterizing ncDNA elements and ncRNA genes.
Cells of Paramecium tetraurelia, a unicellular eukaryote, harbor two types of nuclei with different functions. The diploid micronuclei are mainly transcriptionally silent and undergo meiosis to transmit the genetic information to the next sexual generation. The macronuclei that exhibit a ploidy level of about 1000n express the genetic information and provide the transcriptional templates. The macronuclei develop anew at each sexual generation throughout extensive programmed DNA amplification and rearrangements of a micronuclear genome.
The present assembly of the P. tetraurelia macronuclear genome sequence encompasses 72 Mb, covers >99% of the genome with >99.95% accuracy, and is predicted to harbor 39,642 protein-coding genes from alignments with cDNA sequences and ab initio predictions (Aury et al. 2006). Analysis of the predicted P. tetraurelia proteome showed that 68% of the encoding genes have a duplicate arisen from a recent WGD. Further genome analysis indicated that two older WGDs and a more ancient duplication that might have involved only part of the ancestral genome have shaped the P. tetraurelia genome (Fig. 1). The oldest WGD (OWGD) occurred around the divergence of the Paramecium and Tetrahymena lineages: distances between proteins encoded by P. tetraurelia ohnologs evolved from the OWGD duplicates overlap distances between proteins encoded by P. tetraurelia and T. thermophila orthologs. The more recent WGD (RWGD) took place before the speciation of the P. aurelia taxon into 15 sibling species, and the intermediary WGD (IWGD), during the evolution of the Paramecium phylum into eight taxa. Genes of P. tetraurelia group into ohnolog sets (OHSets) of 1–24 members, depending on how many duplicates have been deleted/maintained throughout evolution. Protein-coding genes from the same metabolic pathway or belonging to the same protein complex have common patterns of loss in P. tetraurelia, while highly expressed genes are over-retained after duplications. This suggested that the retention of protein-coding genes in the P. tetraurelia genome mainly satisfied dosage constraints. Content and evolution of the ncDNA and ncRNA genes of P. tetraurelia has not been investigated yet.
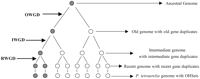
Genome evolution in the Paramecium phylum. Three WGDs occurred during the evolution of the Paramecium phylum (Aury et al. 2006). The OWGD, IWGD, and RWGD gave rise to here named old, intermediate, and recent genomes, respectively. Genes from the ancestral genome gave rise to ohnolog sets (OHSets) of 1–23 members in the P. tetraurelia genome, depending on the timing and number of the deletion and retention events respectively schemed by open and closed circles. In this example, one gene duplicate has been deleted from the old genome following the OWGD and another one from the intermediate genome, following the IWGD. As a consequence the corresponding P. tetraurelia OHSet only includes two members.
Here we have performed a genome-wide analysis of the ncDNA of the P. tetraurelia genome. We first used profile-based methods to recover putative ncRNA genes encoding the 5S rRNAs, snRNAs, snoRNAs, SRP RNAs, and tRNAs. SnoRNA genes were also sought experimentally. Taking advantage of the unique features of the P. tetraurelia genome with regard to the number of WGDs through which it has evolved, we used comparative genomic approaches to analyze ncRNA gene, introns, and intergenic sequences. Finally, we used structure-based methods to screen the intergenic sequences for those that may encode stable and evolutionary conserved RNA secondary structures. Our data provide an extensive and concrete framework for further functional analysis.
RESULTS
ncRNA genes
We sought the genes, the transcription of which produces 5S RNAs, snoRNAs, snRNAs, SRP RNAs, and tRNAs. Besides characterizing the P. tetraurelia genome content for essential ncRNA genes, our aim was to investigate their intron or intergenic localization, identify sequence elements indicative of the transcription machinery they rely on, investigate their evolution and, finally, mark them amongst the ncRNA secondary structures we screened for in the last step of our analysis.
We recovered 417 candidate genes (Table 1; Supplemental Table S1; Supplemental Data). All were inserted within the intergenic DNA except the two snoRNA genes PtCD44-1 and PtCD44-2 that mapped within experimentally recovered introns of genes coding for putative heat-shock 90 proteins (hsp 90). Genes were inserted as singletons (one gene per intergenic DNA segment or intron), but 64 genes that grouped into 20 clusters. Genes within clusters are organized head-to-tail and separated by <1 kb. The U1, U2, U4, and U5 snRNA genes were flanked by a Pol II transcription signature, the U6 snRNA, SRP RNA, and telomerase RNA genes and seven snoRNA gene clusters, by a Pol III signature (Fig. 2A,B).
ncRNA genes in the P. tetraurelia genome

Characterization of ncRNA genes. (A) Pol II-transcribed genes. USEs, TATA boxes, and IEs map upstream of the U1, U2, U4, and U5 snRNA gene 5′ ends, DEs map downstream from the gene 3′ ends. The USE, TATA box, IE, and DE consensus sequences and inter-element distances are shown. (B) Pol III-transcribed genes. USEs and TATA boxes map upstream of the U6 snRNA, SRP RNA, and telomerase RNA gene 5′ ends, polythymidine stretches map at the gene 3′ ends. The USE, TATA box, and polythymidine stretch consensus sequences and interelement distances are shown. (C) PM_01 sequences. USEs, TATA boxes, IEs, and DEs flank the six sequences PM_01. Identical positions between all sequences are shown by stars.
When using the Pol II signature to screen the P. tetraurelia genome, we recovered the sequences PM_01_1 and PM_01_2 of ∼100 bp (Fig. 2C). These sequences have four related sequences in the genome (see below). The Pol III signature did not allow the recovery of any further sequences.
Introns
Annotation of the P. tetraurelia genome predicted >90,000 spliceosomal introns (2.3 introns per gene on average) with an average size of 25 bases. The exon and intron annotations were supported by most EST data (Jaillon et al. 2008). The introns interrupting the >26,000 protein-coding ohnologs that evolved from recent duplicates were aligned and nucleotide identities were calculated, excluding the conserved 5′-GT-3′ and 5′-AG-3′ boundaries (see Materials and Methods). Some 50% of the intron pairs display insertion(s)/deletion(s) [indel(s)] of one, or a few base pairs that were counted as differences in the first step of our analysis (Fig. 3A).

(A) Intron comparison. Identity of intron pairs, (B) pairs of introns differing by 1 bp indels, (C) pairs of introns with no indel, and (D) identity of 4d sites of protein-coding genes evolved from duplicates arisen from the RWGD.
The histogram of nucleotide identity displayed a sprawling distribution suggesting the presence of different classes of introns in the P. tetraurelia genome. Indels could account for up to 15% of sequence divergence between the short introns of P. tetraurelia and be responsible for this distribution. Therefore, we repeated our analysis by splitting introns into two classes according to the presence/absence of indel(s) between ohnologs (Fig. 3B,C). The histograms of nucleotide identity displayed a bimodal distribution in both cases. One distribution appeared binomial, paralleling that of the fourfold degenerated codons (4d sites) (Fig. 3D), the other identifyed an excess of introns with >95% of identity.
Protein-coding gene flanking sequences
Sequences flanking the start and stop codons of the ohnologs arisen from the RWGD were compared by calculating the sequence identity of 50 bp windows slid by 5 bp. Values on the ordinate axis were plotted against window start positions on the abscise axis so that values for positions >0 only reflected ncDNA identity.
A group of genes with low rates of silent substitution of nucleotides, high levels of expression, and ≥5 ohnologs has been reported (Aury et al. 2006). We wondered whether low substitution rates could also characterize sequences flanking some categories of genes and split the sequences into five classes according to the number of expressed sequence tags (EST) with which the adjacent genes associated, 0–2, 3–5, 6–9, 10–15, or ≥16. We noticed two remarkable features. First the gene-flanking sequences from the five classes reached their lowest levels of identity 50 bp downstream from the stop codons for the five classes, and 80 bp and 115 bp upstream of the initiation codons for the first fourth and fifth classes, respectively (Fig. 4A). Second, the higher the number of associated ESTs, the higher the conservation of the 3′ and 5′ sequences within these ncDNA intervals.
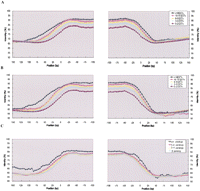
Comparative analysis of gene-flanking sequences. Sequence identity was calculated for windows of 50 bp slipped by 5 bp relatively to the start (left columns) and stop (right columns) codons of genes evolved from recent duplicates. Start and stop codons are indicated by positions 0. (A) Sequences were split into five classes according to the number of associated ESTs: 0-2 (20,406), 3–5 (1,746), 6–9 (689), 10–15 (462), or ≥16 (749) as reported in reference (Aury et al. 2006). (B,C) Intergenic DNA sequences >150 bp were selectively retained for these analyses. Sequences were split into classes according to (B) the number of associated ESTs 0-2 (9,038), 3–5 (712), 6–9 (284), 10–15 (197), and ≥16 (324) or (C) the number of ohnologs in the corresponding OHSets: 2 (5,576), 3–5 (3,980), 5–8 (1,019), or ≥9 (52). Sequences identities reached lower levels 50 bp downstream from stop codons and 80–115 bp upstream of start codons. The higher the number of associated ESTs, the higher the identity of the gene-flanking sequences.
Intergenic DNA in P. tetraurelia is 390-bp long on average but one-third of it is <100 bp. In such short intergenic sequences, the overlapping of the 3′ and 5′ regulatory sequences and gene ends must have biased our analysis. We therefore performed a second analysis of the gene flanking sequences by selecting those mapping within intergenic sequences >150 bp; this length must provide enough space for the specific evolution of the DNA intervals identified above. The >10,000 intergenic sequences were split into five classes accordingly to the EST frequency of the adjacent genes as above. Lowest levels of identity were of 55%–60% in this analysis rather than of 67%. Again, sequence constraints displayed lowest levels 50 bp downstream from stop codons and 80–115 bp upstream of start codons (Fig. 4B). We therefore reached the same conclusion as in our first approach.
Next we split the ncDNA >150 bp into four classes according to the number of ohnologs in the OHSets: 2, 3–4, 5–8, and ≥9. Flanking sequences from the five classes reached their lowest levels of identity as above, 50 bp downstream from the stop codons and 80–115 bp upstream of the initiation codons (Fig. 4C). Higher identity levels of ≈67% characterized sequences mapping upstream of genes of OHSets ≥9.
NcDNA and stable RNA secondary structure conservation
Sequence evolved from recent duplicates were aligned and screened with the Phastcons program so as to recover the most 65% conserved coding sequences (CDSs). While the aligned sequences displayed a wide range of identity, from 45% to 98%, the Phastcons recovered sequences, hereafter named conserved sequences, displayed 84.5% ± 4.5% of identity (Supplemental Fig. S1). CDS coverage of 65% drew out intergenic DNA coverage of 20% (Supplemental Fig. S2).
We then analyzed the conserved intergenic DNA with the RNAz program that captures conserved sequences with conserved stable structures and compare those structures with those of randomized sequences. Using a cutoff of 0.99 for the probability of encoding a stable RNA secondary structure, we characterized 188 sequences of 50–340 bp (Table 2; Supplemental Data). The specificity of this analysis was 80% since only ∼40 sequences could be recovered from random nucleotide assortment from the input sequences. Screening the DNA sequences nonretained by Phastcons but with >50% identity, we identified 40 sequences. Phastcons less-conserved and conserved sequences provided ncRNA structures with a 1:5 ratio even though their abundance displayed a 2:1 ratio. Similar screenings conducted on intergenic ohnologous sequences evolved from the intermediate duplicates allowed the characterization of 11 new sequences.
RNAz predictions
Conserved sequences with conserved stable structures that evolved from the recent or intermediate duplicates identified two PM_01 OHSets including the sequences identified through the Pol II signature and four related sequences (see Discussion), 55 OHSets producing ncRNAs and 74 intergenic DNA sets (see Supplemental Table S1). Finer analysis of the latter mapped one set at the 3′ ends of the MS2 long ncRNA genes that are strongly expressed during sexual reproduction, 5 within ESTs of unannotated regions, 22 within CDSs (wrong annotation of one CDS was responsible for the presence of these sequences), and 46 within intergenic DNAs. Twenty-two of the intergenic DNA motifs mapped within characterized UTRs, while 10 that were adjacent to protein coding gene ends probably defined as yet uncharacterized UTRs. The other 14 intergenic DNA motifs mapped >50 bp from the start or stop codons of the protein-coding gene ends, and may encode regulatory ncRNAs.
DISCUSSION
WGDs provide strong tools for genome-wide insights into the organization, structure, and function of ncDNA elements and ncRNA genes. Three successive WGDs and an ancient duplication have marked the evolution of the Paramecium phylum. Phylogenetic relationships can be established between genes clearly enough to group them into OHSets of 1–24 members, depending on how many duplicates have been deleted/maintained throughout evolution. Here we have used these facts to analyze the ncRNA genes, spliceosomal introns and intergenic DNA of the P. tetraurelia genome.
Purifying selection: One main actor of ncRNA evolution in Paramecium
We looked at five key classes of ncRNA and characterized 417 putative ncRNA genes. Additional experimental screenings will allow further ncRNA genes to be recovered, especially for those producing snoRNAs (see Supplemental Data) and tRNAs (see below). The genes that we characterized are expected however to produce a large fraction of the 5S rRNAs, snRNAs, snoRNAs, SRP RNAs, and tRNAs. These genes are known to evolve through high constraints in all kingdoms. ncRNA ohnologs indeed display high sequence identity in the P. tetraurelia genome (>80% of coverage in our phastcons analysis; see Materials and Methods). We investigated the molecular mechanisms of their evolution.
Purifying selection and snRNAs
SnRNAs are crucial components of spliceosomes that remove introns from primary transcripts.
The U6 snRNA genes are flanked by identical regulatory sequence elements in P. tetraurelia, T. thermophila, A. thaliana, and vertebrates (see Fig. 2).
The U1-2 and U4-5 snRNA genes however display differences in the 5′ region that harbors an upstream sequence element (USE) in vertebrates (Jawdekar and Henry 2008), an USE and a TATA-like box separated by 33-34 bp in A. thaliana (Waibel and Filipowicz 1990) or by 18 bp in P. tetraurelia (see Fig. 2) and T. thermophila (Supplemental Fig. S3; Orum et al. 1992). Differences in the distances separating the USE and the TATA-like box upstream of the U1-2 and U4-5 snRNA genes between ciliates and plants might reflect differences in the occupancy of the promoter regions by transcription factors. In any case, the regulatory sequence elements that flank these snRNA genes of ciliates appear more closely related to those of plants than to those of vertebrates.
SnRNAs are usually encoded by moderately repeated genes that show little variation within a given genome. The two U4 snRNA genes have evolved from a pair of recent gene duplicates, while the three U5 or U6 snRNA genes have evolved from two pairs. The U4, U5, or U6 snRNA genes only display WGD relationships, therefore evolved from one ancestor gene and group into 1 OHSet. The four U2 snRNA genes have evolved from two pairs of recent gene duplicates that map onto two DNA ohnologous blocks, each of which harbors two genes separated by 70 kb. The organization of the U2 snRNA genes may reflect a cis duplication of an intermediate duplicate. In contrast the four U1 snRNA genes group into two OHSets.
High levels of identity (>90%) characterize the the genes and upstream 70 bp in five of the seven pairs of snRNA ohnologs having evolved from recent duplicates (75% of identity characterize the upstream 70 bp in a sixth pair, while the snRNA ohnologs of the seventh pair lay <50 bp from the neighboring genes so that sequence identity upstream of the genes could not be analyzed). The high levels of sequence identity of the P. tetraurelia snRNA ohnologs and of the base pairs that are adjacent to the transcript ends may attest conversion events, the propagation of which had been stopped by physical constraints. Lower levels of identity (∼75%) however characterize the 70 bp mapping of the snRNA ohnologs that evolved from intermediate duplicates although the genes are 99% identical. This strongly suggests that purifying selection rather than conversion has been one main actor in the evolution of the P. tetraurelia snRNA genes and flanking base pairs.
Purifying selection and snoRNAs
Most characterized snoRNAs of eukaryotes and archebacteria guide modifications of the rRNAs and snRNAs that are thought to be involved in RNA folding as well as in RNA–RNA and RNA–protein interactions. Modifications of rRNAs and snRNAs are achieved through interactions of RNA molecules and snoRNA short guide sequences of few bases (see Materials and Methods). In P. tetraurelia, the 19 box H/ACA OHSets are expected to promote the pseudouridylation of 2 snRNA and 27 rRNA sites, and the 48 box C/D OHSets, the methylation of 3 snRNA and 51 rRNA sites (note that some OHSets harbor members with two guide sequences and that snoRNA ohnologs guide identical modification[s], see below).
Orphan snoRNAs are snoRNAs without any base complementarity to any rRNA or snRNA. Identification of orphan targets is a difficult task because of the large number of candidates but showed surprises. In human, snoRNAs HBII-52 control the editing and alternative splicing of the serotonin receptor 5-HT(2C)R mRNA (Cavaille et al. 2000; Kishore and Stamm 2006), while a lack in HBII-85 snoRNAs plays a causal role in the Prader-Willi syndrome of mental retardation (Sahoo et al. 2008). Interestingly, recent work identified orphan and nonorphan snoRNAs as novel precursors of miRNAs (Ender et al. 2008; Saraiya and Wang 2008). In P. tetraurelia, we could recover four putative orphan snoRNA OHSets that require further analysis.
SnoRNA-guided modifications differ between eukaryotes. This and the fact that snoRNA genes can behave as mobile elements (Weber 2006) suggested that part of snoRNA duplicates may be exempt from evolutionary constraints, mutate and guide new RNA modifications. The P. tetraurelia genome provides an unusual opportunity to test this hypothesis since 129 out of 142 snoRNA genes group into 58 OHSets of size ≥2 into which duplicate evolution can be analyzed. Box H/ACA sequence guides and box C/D guide cores are identical between ohnologs evolved from recent or intermediates duplicates in all OHSets, but two in which sequences harbor one difference (see Supplemental Data).
Different levels of nucleotide identity however characterize the box H/ACA and box C/D snoRNA ohnologs evolved from recent (96.7% ± 3.6%) or intermediates (83.6% ± 9.6%) duplicates. These strongly suggest that snoRNA genes evolved through purifying selection in P. tetraurelia. This purifying selection acted strongly on all the ohnologs evolved from recent or intermediate duplicates, leaving few chances for new guiding functions to arise within the corresponding molecular times.
Eighteen snoRNA OHSets group into clusters. OHSets PtCD07 and PtCD08 probably evolved from a cis duplication since their guide cores differ by an indel of 1 bp (see Supplemental Fig. S6). OHSets PtCD01-06 have different guide cores as do OHSets PtCD09-10, PtCD11-12, PtCD13-14, PtCD40-41, or PtCD42-43. The observation we made that purifying selection maintains identical cores between snoRNA ohnologs arisen from intermediary or recent duplicates strongly suggests that, if cis duplications are responsible for these clusters, they must have taken place within the old genome, before the IWGD. Alternatively, gene horizontal transfer within the old genome may have been at the origin of these clusters.
Purifying selection, 5S RNAs and tRNAs
The P. tetraurelia anticodon repertoire harbors 44 anticodons complementary to the 61 universal sense codons and 2 anticodons for the specific decoding of the 5′-TAG-3′ and 5′-TAA-3′ codons. No Sec tRNA genes were recovered. The identification of Sec tRNA genes requires specific computational analysis and cloning of the predicted products as has been done in T. thermophila (Shrimali et al. 2005). The selenophosphate synthetase 2 (SPS2) protein is essential to generate the selenium donor for selenocysteine biosynthesis (Xu et al. 2007). The P. tetraurelia genome harbors a gene encoding a putative SPS protein, suggesting it also encodes Sec tRNAs.
Seventeen of the tRNA OHSets group into OHSets of size ≥3 into which the levels of identity of the genes evolved from recent and intermediate duplicates can be compared. Ten of the tRNA OHSets harbor identical members but seven harbor one member of lower identity (∼85%). On the other hand, all 5S RNA genes display identical sequences but seven that group into three OHSet and have 1–4 nucleotide (nt) differences when compared to the former. If conversion events are responsible for sequence homogeneization between tRNA OHSet members or 5S RNA genes, they have been rare enough not to involve all of them. This suggests that purifying selection has been the main actor of sequence homogenization between members of the tRNA and 5S RNA OHSet. In that hypothesis, the tRNA OHSet members of lower identity may be nonfunctional members no more submitted to any evolutionary selection.
Death: Another main actor of ncRNA evolution in Paramecium
The 417 ncRNA genes group into 210 OHSets (see Table 1). Although the OWGD, IWGD and RWGD might have led to OHSets of eight members, no OHSet has a size ≥6 and only two have a size of five. Furthermore, only two OHSets harbor members arisen from the two old gene duplicates. The strict requirement in 5S rRNAs, snRNAs, snoRNAs, SRP RNAs, and tRNAs of cells implies that the Paramecium ancestral genome harbored an ancestral gene for most if not all ncRNA OHSets of P. tetraurelia, and that one of the duplicates arisen following the OWGD was lost in all but two cases.
Forty-two and 160 OHSets contain members arisen from two and one copies of the intermediate genome, respectively (not including genes apparently arisen from cis duplication). Sixty-four and 94 OHSets contain members arisen from one and two copies of the recent genome, respectively. Some OHSets of size ≤2 may have arisen from segmental duplications in the recent genome. Loss of ncRNA genes following the IWGD and RWGD however concerned ∼80% and ∼40% of the intermediate and recent duplicates, respectively. Telomerase RNA duplicates, in particular, have been submitted to repetitive deletions since the genome only harbors one gene (McCormick-Graham and Romero 1996) and no related sequence. The telomerase RNA gene is present as a single, or very limited number of copies in all eukaryote genomes suggesting that deletion, but not purifying selection, is able to ensure a qualitative and quantitative control of telomerase RNAs.
Short introns distribute into two classes in P. tetraurelia
A distinct population of short introns has been characterized in organisms as diverse as A. thaliana, C. elegans, D. melanogaster, human, and S. cerevisiae (Lim and Burge 2001). Short introns have maximal lengths specific in each organism, ranging from 60 to 134 bp in the first four organisms, and of 191 bp S. cerevisiae. They represent about half of the whole intron population in these organisms except in human where they are only 10%. Short introns are recognized by the splicing machinery via splice site pairings across introns rather than exons. This mode of recognition (intron definition model) was initially proposed because mutations of 5′ splice sites of short introns lead to intron retention rather than to exon skipping, and later confirmed by splice site coevolution (Berget 1995; Lim and Burge 2001).
Comparative genomics is a complementary approach for identifying sequence elements involved in intron recognition. To our knowledge, comparative genomics of introns has been developed on a genome-wide scale only between humans and chimpanzees, and D. melanogaster and simulans (Halligan and Keightley 2006; Gazave et al. 2007). Comparative genomics was also conducted on 605 pairs of introns resulting from a recent segmental duplication in the rice genome (Guo et al. 2007). Intron divergence negatively correlated with intron length in all cases.
All the P. tetraurelia introns are probably excised from the pre-RNAs by U2-type spliceosomes. We could not recover any U11- or U12-snRNA genes, confirming previous reports (Davila Lopez et al. 2008), nor any associated proteins, (L. Amar, unpubl.). The P. tetraurelia genome harbors two ohnologous genes encoding putative debranching enzymes suggesting that the encoded introns form lariats.
We used comparative genomics to investigate whether recognition of the P. tetraurelia short introns follows the rules depicted for other eukaryotes. Short introns largely predominate within the P. tetraurelia genome with >96% of them being 20–34 nt in length (Jaillon et al. 2008). Insertion of snoRNA genes is responsable for the unusually long introns of 180 bp of the hsp 90-like genes.
A large majority (90%) of the pairs of the introns that are inserted within the ohnologs arisen from recent duplicates display indels of one or a few base pairs and low levels of identity. Recognition of these introns may involve a major role of the conserved splice sites. A minor but still abundant fraction of the P. tetraurelia intron pairs (10%) displays a high level of sequence identity (>95%) attesting slow if any sequence evolution. Sequence identity can extend into 30 bp of the adjacent exons that do not display any difference even though an average of 27% of difference is expected at 4d sites. Previous comparative genomics of introns mainly focused on evolutionary differences between short and long introns. Short introns were treated as one indistinct population in the primate analysis, impeding finer analysis of the levels of identities. Genomes of Drosophila and rice however do display large differences in the level of sequence identity of the short ohnologous introns suggesting the presence of a distinct class of short introns in these genomes.
Conserved and ultraconserved elements of >100 bp and >200 bp, respectively, have been identified between introns, or intron–exon regions, of human, rat, and mouse. These elements may define splicing regulatory elements, especially in cases of alternative splicing leading to premature stop codons and non-sense-mediated decay (NMD) (Bejerano et al. 2004; Katzman et al. 2007; Ni et al. 2007). Enrichment of conserved elements within the RNA Recognition Motif of RNA binding domains and splicing regulators suggested that these elements participate in maintaining a tightly tuned homeostasis of RNA-binding protein levels in vertebrate cells. Alternative splicing is very limited in P. tetraurelia: <0.9% of the ∼14,000 introns covered by at least two ESTs were found to use alternative splice sites, usually closely spaced 3′ sites (Jaillon et al. 2008). Study of intron excision in cells deficient for the NMD pathway showed that introns are excised with a wide range of efficiency (Jaillon et al. 2008). As a consequence, stop codon-harboring introns lead the RNA molecules until they are retained to degradation through the NMD pathway. Sequence conservation within introns may participate to define constitutive excision efficiencies in P. tetraurelia, and thereby participate in fine tuning the level of some mRNAs.
Sequences flanking the highly expressed genes evolve more slowly
At gene 3′ ends, sequences evolved from recent duplicates display decreasing conservation within 50 bp, a DNA interval which size is close to that of the cloned 3′ UTRs (52 bases). Upstream of the gene start codons, sequences evolved from recent duplicates display decreasing conservation within 80–115 bp, a DNA interval, where its size is slightly greater than the average size of the cloned 5′ UTRs (58 bases), and may therefore harbor transcription signals. About 48% of the intergenic DNA is <150 bp in the P. tetraurelia, while ∼20% of the intergenic DNA is <50 bp. In these cases, transcription signals might overlap with gene coding ends.
Saccharomyces genomes are compact genomes with an average length of the intergenic DNA of 500 bp, the organization of which can be compared to that of P. tetraurelia (Nagalakshmi et al. 2008). In the Saccharomyces genomes, the initiation and termination gene codons are first followed by sequences of low identities, then by a faint enhancement of sequence conservation 75–125 bp downstream from the latter or by a peak of conservation, 125–250 bp upstream of the former (Cliften et al. 2003). This organization has been interpreted as a consequence of a spatial restriction between regulatory sequences and gene ends. Gene-flanking sequences display evolutionary differences between the compact genomes of P. tetraurelia and Saccharomyces species indicating different regulations of the transcription and/or translation processes.
A distinct evolution characterizes the gene-flanking sequences harboring a TATA box in Saccharomyces. These sequences have a broader and higher peak of conservation. Genes with high expression levels or from OHSets of size ≥9 display enhanced conservation of the flanking sequences in P. tetraurelia (see Fig. 3C). The promoters of these genes may contain more regulatory sequences.
Interplay of ncRNA and protein-coding genes in Paramecium
The massive loss of duplicates following the OWGD and the frequent loss of duplicates that followed the IWGD and RWGD resulted in shrunk sets of 1–2 ncRNA ohnologs in P. tetraurelia. The P. tetraurelia genome encodes twofold more genes (≈40,000 genes) than the ancestral Paramecium genome (≈20,000 genes) (Aury et al. 2006). The evolutionary pattern of the ncRNA genes paralleled that of the protein-coding genes. The efficiencies of the transcription or translation processes, in which a large fraction of the ncRNA genes participates, have probably been maintained throughout the evolution of the Paramecium phylum.
The P. tetraurelia genome encodes two-, three-, and sevenfold less snRNA, tRNA, and 5S RNA genes, respectively, than the genome of T. thermophila (Eisen et al. 2006). The ciliates P. tetraurelia and T. thermophila are expected to have similar cell metabolism requirements, so that if most ncRNA genes are housekeeping genes, differences in their numbers might reflect differences in their transcription efficiency between both ciliates. Data on gene expression and cell metabolism are required to allow insights into the consequence of the different ncRNA gene organization in the model eukaryotic genomes.
Using the RNAz program, we identified 46 evolutionary sets of RNA secondary structures from the intergenic DNA. These putative transcripts differ in size and structure from the scanRNAs of 27–30 nt that are produced from the micronuclear genome during meiosis (Lepere et al. 2008). In contrast with the Evofold program, the RNAz program preferentially recovers GC-rich structures so that we may have missed RNA secondary structures of the P. tetraurelia A+T-rich ncDNA. However, the Evofold program requires more aligned sequences than those present within the RWGD (two sequences) and IWGD (four sequences) genomic subsets.
RNA secondary structures may be functional as regulatory elements of the MS2 long ncRNA and 32 mRNAs to which they are adjacent. NcRNA transcripts of ≈100 bp with stable and conserved secondary structures are also expected for 14 DNA motifs that we recovered, >50 bp distant from any CDS. Amongst the latter are the six PM_01 sequences that are flanked by a typical Pol II transcription signature. The corresponding ncRNAs, if they exist, may participate to the recognition of some of the short introns in the pre-mRNAs. These ncRNAs may also participate to the recognition of the ∼50,000 internal eliminated sequences (IESs) that are precisely excised from the P. tetraurelia micronuclear genome in the course of macronuclear genome development through still unknown mechanisms.
MATERIALS AND METHODS
DNA ohnologous block alignments
The P. tetraurelia macronuclear genome sequence version 2.1 and its February 2007 annotation were downloaded from the Genoscope Paramecium genome browser (http://www.genoscope.cns.fr/externe/Francais/Projets/Projet_FN/). The DNA ohnologous blocks were defined on the basis of protein-encoding gene synteny by intragenome comparison (Aury et al. 2006). The DNA ohnologous blocks having arisen from the AD, OWGD, IWGD, and RWGD were grouped within genome subsets. Sequence alignments were performed with the program MULTIZ (Blanchette et al. 2004).
NcRNA gene identification
As a first step, we used the BLAST program to identify intergenic sequence elements homologous to conserved profiles of the Rfam database version 8.0 (http://www.sanger.ac.uk/Software/Rfam/index.shtml) (E value cutoff of 1) (Altschul et al. 1997; Griffiths-Jones et al. 2005). In the case of multihits with the Rfam database, the one with the lowest BLAST E value was kept. Then, we used the Infernal program to check the candidates (Eddy 2002). The candidates with an Infernal score >10 bits and a Infernal E value < 0.01 were considered as ncRNA gene candidates. Intergenic sequences were also scanned with the Erpin program (Gautheret and Lambert 2001). The Erpin batch scripts and profiles were obtained from the Erpin online server (http://rna.igmors.u-psud.fr/erpin/). Hits with E values <0.01 were kept as ncRNA gene candidates. tRNAscan-SE (Lowe and Eddy 1997) was specifically used for the recovery of tRNA genes. tRNAscan-SE provided tRNA gene candidates and putatively nonfunctional related sequences because of their poor primary or secondary structure. Supplemental Figure S4 displays a graphic representation of the ncRNA gene candidates that were recovered with the different programs. Manual editing of the results led to the elimination of obvious false positives. The P. tetraurelia genome was also screened for loci harboring the regulatory elements associated with Pol II transcription.
SnoRNA gene identification
Box C/D snoRNAs mainly guide ribose 2′-O-methylations, while box H/ACA snoRNAs guide conversions of uridines into pseudouridines. SnoRNA genes were identified using the snoRNA Mining Platform (snoRMP) based on the SnoScan and SnoGPS algorithms, combined with secondary structure prediction and comparative genome analysis (Lowe and Eddy 1999; Chen et al. 2003; Schattner et al. 2006; Chen et al. 2008). The sequences of the P. tetraurelia rRNAs and snRNAs were used for the identification of the snoRNA complementary guiding sequences. Interspecies comparisons suggested that the published P. tetraurelia rRNA sequence (GenBank Accession Number AF149979) is incomplete. Using the raw genome sequencing data, we indeed assembled an rRNA sequence that is 200 bp longer (GenBank Accession Number EU828456). The box C/D snoRNA search program identified segments <150 bp that include box(es) C (RUGAUGA), box(es) D (CUGA), at least 10 nt complementary to a rRNA or snRNA sequence and terminal short inverted repeats. Among these five elements, we considered primarily the quality of the potential snoRNA-rRNA duplexes: the duplex length, the number and position of the mismatches in the core sequence, and G:U pairings. Guiding core sequences extend from the third to the 11th positions upstream of the D box with the fifth nucleotide invariably pairing to the modified nucleotide (fifth nucleotide rule) (Kiss-Laszlo et al. 1996; Chen et al. 2007). The box H/ACA snoRNA search program identified segments <200 bp exhibiting a typical hairpin–hinge–hairpin–tail secondary structure, a box H (ANANNA) in the hinge region a box ACA (ACA or AUA) in the tail, as well as two sequences of the pocket of the hairpin complementary to a rRNA or snRNA sequence (each >3 nt and the sum >9 nt). Since snoRNA genes often cluster in eukaryote genomes, sequences flanking the snoRNA candidates were examined for additional snoRNA genes. BLAST searches were performed with each snoRNA candidate to characterize homologous sequences within the whole genome.
CDNA cloning
cDNA libraries were constructed as previously described with some modifications (Huang et al. 2005). Briefly, about 25 μg of total P. tetraurelia RNA was polydenylated using poly(A) polymerase (Takara). Then poly (A)-tailed RNA was reverse-transcribed into first strand of cDNA in a 20 μL reaction mix containing 200 U of MMLV reverse transcriptase (Promega) and 0.2 μg box D-anchor primers dT16-CD. The reaction mixture was size-fractioned on a denaturing 8% polyacrylamide gel (8M urea and 1X TBE buffer). cDNAs with sizes ranging from 60 to 120 nt were excised, eluted from the gel; and tailed with poly(dG) at the 3′-end by using terminal deoxynucleotidyl transferase (Takara). Poly(G)-tailed cDNA was amplified by primers PolyCM or PcTGA and dT16-CD to construct box C/D snocDNA enriched libraries. Then amplified fragments of PCR products sized 110–170 were gel eluted using a QIAquick PCR Purification Kit (Qiagen), cloned into plasmid pMD18T (Takara) and transformed into Escherichia coli DH5. The cDNA libraries were screened by PCR with the P47 and P48 universal primer pair. Only the recombinant plasmids carrying fragments of expected size were selected to be sequenced. Out of the 244 sequences we obtained, 4 matched rDNAs from other species than P. tetraurelia, 16 matched P. tetraurelia snDNAs, 41 matched P. tetraurelia rDNAs, and 183 (75%), putative snoDNAs. Oligonucleotides were synthesized and purified by Biosune Co. (Shanghai, China);
-
dT16-CD, 5′-TTTTTTTTTTTTTTTTNNNNTCAG-3′;
-
PolyCM, 5′-GGAATTCGGATCCCCCCCCCCCCCCCC-3′;
-
PcTGA, 5′-CATTGGATCCCCCCCCCCCCCCCCCCNNNNNNNTGATGA-3′;
-
P47, 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′; and
-
P48, 5′-GAGCGGATAACAATTTCACACAGG-3′.
Conserved sequence identification
Conserved sequences were identified with the program PhastCons of the PHAST package that is based on a two-state phylogenetic hidden Markov model (phylo-HMM) for conserved and nonconserved states (Siepel et al. 2005). The degree of “smoothing” of the phylo-HMM was constrained by forcing the phylogenetic information threshold (PIT) calculated by consEntropy of the PHAST package to be equal to 9.8 bits for all sequence sets. The choice of these parameters defined minimal conservation lengths (Lmin) of 12 bp, 25 bp, and 100 bp between the ohnologous sequences arose from the OWGD, IWGD, and RWGD, respectively (Supplemental Table S2). Lmin differences between paralog comparisons reflected differences in the number and in the evolutionary distance of the aligned sequences. In the case of the ohnologous sequences arising from the ancient duplication, the PIT value and Lmin could not be determined because of the high number of possible sequence comparisons (up to 24 comparisons). Parameters “target coverage” and “expected length” defined the percent of the CDSs conserved and expected length of the conserved sequence fragment within sequence alignments. The nonconserved trees were estimated by 4d sites in coding regions with the phyloFit program also from the PHAST package, and the conserved trees were estimated from the genome-wide sequence alignments by phastCons program, for each pair of target coverage and expected length. The same parameters were used to generate base-by-base conservation scores for each alignment. All base-level coverage statistics were computed using the featureBits program of the UCSC Genome Browser package (http://www.soe.ucsc.edu/∼kent/src/unzipped/hg/featureBits). Assignment of the conserved sequences into CDSs, introns or intergenic DNA was based on the annotation of the Paramecium genome (http://www.genoscope.cns.fr/externe/Francais/Projets/Projet_FN/) (see Supplemental Data).
Conserved structure identification
The sequence alignments of the intergenic sequences were extracted by the mafFrags program of the UCSC Genome Browser package from sequence alignments of the IWGD and RWGD genomic subsets. Extracted alignments longer than 50 bp and with mean pairwise identities higher than 50%, were scanned for conserved stable structures with the RNAz program on overlapping windows of size 120 bp and slide 50 bp, using cutoffs of P = 0.5, 0.9, or 0.99 (Washietl et al. 2005). Overlapping hits were combined to a single genomic region (“locus”). The program rnazRandomizeAln.pl of the RNAz package was used to generate 100 randomized sequence alignments by shuffling the alignments of the original data set by columns (see Supplemental Data).
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org. ncRNA gene annotation and sequence conservation tracks can be obtained at the Paramecium DataBank (http://paramecium.cgm.cnrs-gif.fr) (Arnaiz et al., 2007). An example of the conservation tracks is shown in Supplemental Figure S5.
ACKNOWLEDGMENTS
We thank Lenny Rabinow and Marie-Laure Samson for the critical reading of the manuscript, Daniel Gautheret for the new Erpin script package and the anonymous referee that took time to improve the quality of our manuscript. This research is supported by the National Natural Science Foundation of China (key projects 30830066 and 30870530), the funds from the National Basic Research Program (No. 2005CB724600), and those from the Association Franco-Chinoise pour la Recherche Scientifique et Technique (PRAE99-02). C-L.C. was a recipient of grants from the French government (French Embassy Ph.D. program) and the Excellent Doctor Thesis program of the Sun Yat-Sen University.
Footnotes
-
↵4 Present address: Centre National de la Recherche Scientifique (CNRS), UPR 2167, CGM, Gif sur Yvette, 91198, France.
-
Reprint requests to: Laurence Amar, Institut de Biologie Animale Intégrative et Cellulaire, Université Paris Sud, Orsay 91405, France; e-mail: laurence.amar{at}u-psud.fr; fax: 33 1 69 15 49 49; or Liang-Hu Qu, Key Laboratory of Gene Engineering of the Ministry of Education, State Key Laboratory for Biocontrol, Zhongshan University, Guangzhou, 510275, People's Republic of China; e-mail: lssqlh{at}mail.sysu.edu.cn.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1306009.
-
- Received August 8, 2008.
- Accepted December 23, 2008.
- Copyright © 2009 RNA Society








