
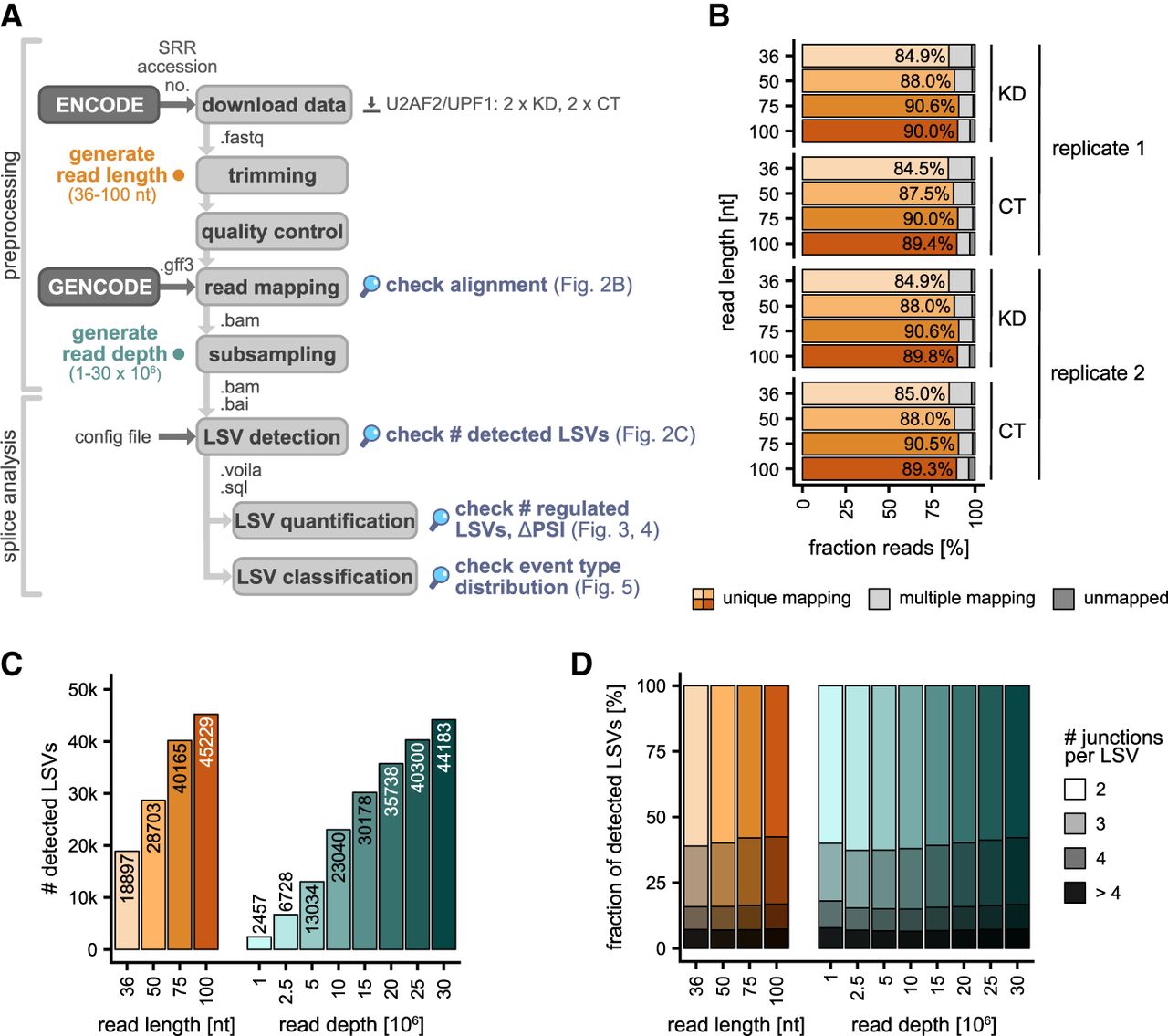
Longer reads and a higher read depth allow for better detection of LSVs. (A) Workflow of preprocessing and AS analysis. First, the raw sequencing data were downloaded from ENCODE (fastq-dump), trimmed (Trimmomatic), checked for quality (FastQC), and then mapped to the human genome (STAR) with GENCODE gene annotation. Subsampling the resulting BAM files simulated different sequencing depths (samtools view). AS analysis was performed using MAJIQ. Workflow includes work steps (light gray boxes), databases (dark gray boxes), and file formats (next to arrows). Colored bullets indicate in silico variation in read length (orange) and sequencing depth (sea green). Magnifiers mark stages where the influence of RNA-seq characteristics was tested. (B) Longer reads show slightly better mappability. Proportion of uniquely mapped, multimapped, and unmapped reads for four different read lengths (36–100 nt, light to dark orange) of the KD and control replicates of the U2AF2 KD experiment. (C) Number of LSVs detected by MAJIQ for different read lengths (36–100 nt, light to dark orange) and read depths (1–30 M reads, light to dark sea green). (D) LSV complexity. Fraction of LSVs with a given number of junctions therein for different read lengths (36–100 nt, light to dark orange) and read depths (1–30 M reads, light to dark sea green).








