
Progress and challenges in profiling protein–RNA and protein-associated RNA–RNA interactions
- Therapeutic Innovation Center and the Verna Marrs McLean Department of Biochemistry and Molecular Pharmacology, Baylor College of Medicine, Houston, Texas 77030, USA
- Corresponding author: eric.vannostrand{at}bcm.edu
Abstract
RNA binding proteins (RBPs) play essential roles in post-transcriptional gene regulation by interacting with a wide range of RNA targets. In addition to regulating RNA processing via individual RBP–RNA interactions, there is a growing appreciation of the regulatory impact of protein-associated RNA–RNA interactions that include both well-studied examples of small regulatory RNAs (e.g., microRNAs, snRNAs, snoRNAs, piRNAs) guiding ribonucleoprotein complexes to their targets as well as structured RNA elements defining the interaction landscape for an RBP. To elucidate the full scope of RBP–RNA interactions, CLIP (crosslinking and immunoprecipitation)-based methods have emerged as powerful tools. Even with the wide application of CLIP and variant approaches, these methods are still under significant ongoing advancement to better accommodate diverse biological systems and experimental demands and improve scalability. In particular, recent years have seen a focus on improved techniques to globally profile protein-associated RNA–RNA interactions. In this review, we provide a summary of recent improvements in traditional CLIP methods that improve the mapping of RBP–RNA interactions, with particular focus on those that specifically enable the profiling of protein-associated RNA–RNA interactions. We discuss the unique challenges involved in mapping protein-associated RNA–RNA interactions and highlight different ways current approaches address these challenges in order to offer a practical framework for researchers seeking to investigate RBP-associated RNA interactions.
Keywords
INTRODUCTION
RNA molecules serve as key regulators of diverse cellular processes, governed by a complex network of molecular interactions. Among these, RNA-binding proteins (RBPs) represent central regulators, orchestrating RNA maturation, modifications, and subcellular localization through interacting with RNA molecules (Hentze et al. 2018; Van Nostrand et al. 2020a; Kelaini et al. 2021; Fagre and Gilbert 2024). In parallel, recent advances have highlighted the importance of RNA–RNA interactions (RRIs), particularly those stabilized or mediated by RBPs. Intramolecular RRIs, which occur within the same RNA molecule, are fundamental to the formation of RNA structures that have emerged as key regulatory elements in controlling RNA translation, splicing, stability, and function (Anokhina et al. 2013; Hansen et al. 2013; Yang et al. 2021); intermolecular RRIs, formed between distinct RNAs, enable regulatory RNAs to control the expression and functions of target RNA transcripts (Kung et al. 2013; Iwasaki et al. 2015; Huang et al. 2022b; Diener et al. 2023). Some RRIs, such as riboswitches and ribozymes, can function independently of proteins (Serganov and Patel 2007). However, many occur and act as part of ribonucleoprotein (RNP) complexes—RNA structure can facilitate (or block) RBP association, while RBP binding can similarly promote (or hinder) RNA–RNA interactions.
Moreover, certain RNA–RNA interactions act as molecular recognition elements, enabling the regulatory activity of an associated RNP complex. This latter group includes many well-studied classes of regulatory RNA that drive control of gene expression: microRNAs act as guides to recruit the RISC complex to induce RNA degradation and inhibit translation (Iwakawa and Tomari 2022; Diener et al. 2023; Shang et al. 2023); small nucleolar RNAs (snoRNAs) guide Fibrillarin to catalyze 2′-O-methylation or Dyskerin to perform pseudouridylation of rRNA, snRNA, mRNA, and other RNAs to alter their binding and function (Tycowski et al. 1998; Kiss 2002; Zhang et al. 2023; Liu et al. 2025); spliceosomal small nuclear RNAs (snRNAs) participate in the recognition of splice sites to enable the spliceosome to properly excise introns during pre-mRNA splicing (Martínez-Lumbreras et al. 2024); and a variety of other small RNAs including piRNAs, 22G-RNAs, and sRNAs play roles as guide RNAs for RNP complexes in species ranging from human to Escherichia coli (Grivna et al. 2006; Gu et al. 2009; Rodgers et al. 2023). RBP–RNA interactions are highly dynamic and context-dependent and can vary across cellular compartments, cell types, and pathological conditions (Gebauer et al. 2021; Kelaini et al. 2021), and we have only begun to understand the complex roles these RBP-mediated RNA interactions play in human gene regulation and physiology.
Transcriptome-wide identification of RNA binding protein interactions, particularly RBP-mediated RNA–RNA interactions, has advanced our understanding of this field. A variety of techniques have been developed to characterize RBP–RNA interactions in vivo (Licatalosi et al. 2008; Hafner et al. 2010; König et al. 2010; Van Nostrand et al. 2016; Zarnegar et al. 2016). Building upon established RBP–RNA profiling strategies, protein-based RRI mapping has emerged as a powerful tool to capture RNA duplexes in vivo (Helwak et al. 2013; Moore et al. 2015; Sugimoto et al. 2015; Nguyen et al. 2016; Song et al. 2020; Manakov et al. 2022). These approaches substantially advance our understanding of regulatory roles and mechanisms of the RNP complex generally as well as the specific biological function of individual guide RNAs.
In this review, we aim to provide an overview of techniques for profiling protein–RNA interactions, focusing on recent modifications that have not been described in extensive recent reviews (Lee and Ule 2018; Wheeler et al. 2018; Hafner et al. 2021), with a particular focus on approaches that map RBP-associated RNA–RNA interactions. Building upon recent comprehensive overviews of techniques to map RNA–RNA interactions and characterized functions of these RRIs in RNA biogenesis in vitro, in vivo, and in situ (Singh et al. 2022; Ye et al. 2024), here we provide a comparative overview of the key experimental workflows and analytical strategies employed in RRI technologies and discuss the strengths and challenges of the current methods, providing an introduction to ongoing work in this burgeoning frontier in RNA biology.
MAPPING PROTEIN–RNA INTERACTIONS WITH CLIP AND RELATED TECHNIQUES
Originally, RIP (RNA immunoprecipitation) was employed to capture native RBP–RNA interactions. In this approach, RBP–RNA complexes were precipitated using a protein-specific antibody, and then either coupled with northern blotting or qPCR (for low-throughput assays) or microarrays or high-throughput sequencing (for transcriptome-wide profiling) (Tenenbaum et al. 2000; Keene et al. 2006; Zhao et al. 2010). While RIP has contributed valuable insights into RBP-associated RNA populations, early versions lacked the resolution to pinpoint the precise binding sites of an RBP. In addition, due to the lack of crosslinking in early variants to covalently stabilize RBP–RNA interactions, it is possible that the RBP may dissociate from RNA during the procedure (or even that new interactions may form during the lysis or immunoprecipitation). To overcome these limitations, CLIP (crosslinking and immunoprecipitation) was developed (Ule et al. 2003, 2005; Licatalosi et al. 2008). In CLIP (Fig. 1, left), cells are exposed to ultraviolet (UV) irradiation to covalently crosslink RBPs to their directly bound RNA targets, thereby stabilizing in vivo interactions. Following cell lysis and RNA fragmentation, the protein–RNA complexes are immunoprecipitated, and 3′ adaptors are ligated to the RNA. The complexes are then purified by SDS-PAGE and transferred to a nitrocellulose membrane, as non-crosslinked RNA does not transfer to nitrocellulose and thus only RNA crosslinked to protein of the desired size can be specifically excised and isolated. After protein digestion, RNA is reverse transcribed into cDNA for sequencing. This approach combined with high-throughput sequencing enabled transcriptome-wide mapping of RBP binding sites and has become a foundational technique in the study of protein–RNA interactions (Darnell 2010).

Overview of key steps of protein-dependent and protein-independent methods to detect protein–RNA or RNA–RNA interactions. Schematic of differential core steps in (left) CLIP and its variant protocols, (center) RBP-associated RRI detection protocols, and (right) direct RRI detection protocols.
A number of modified CLIP approaches have been developed over the past two decades to further improve the efficiency and specificity of RBP–RNA interaction profiling, which have recently been extensively reviewed (Lee and Ule 2018; Wheeler et al. 2018; Hafner et al. 2021). These modifications have focused on various directions for distinct applications, including modifying the enzymatic steps to increase efficiency of converting immunoprecipitated RNA into the final sequencing library, modifying crosslinking to increase yield of recovered RNA (particularly for RBPs that weakly or do not bind single-strand RNA and are thus refractory to standard UV crosslinking), modifying how enrichment of the targeted RBP is performed (including non-antibody options that enable more stringent washes to remove additional background signal), and even more dramatic changes that enable RBPome-scale profiling in a single experiment.
Library preparation improvements
Most current CLIP methods make use of reverse transcription termination at the RBP-crosslinked nucleotide, which was first utilized in iCLIP (individual-nucleotide resolution UV crosslinking and immunoprecipitation) to not only enable individual nucleotide resolution but also improve library yield (by ligating the second 5′ adaptor after reverse transcription, terminated cDNA products are no longer lost) (König et al. 2010). More recent improvements include further optimizations. irCLIP (infrared CLIP) replaces radioactive labeling with infrared dye (IR800) and biotin-conjugated adaptors, enhancing the detection and adaptor ligation caused by radiolabeling (Zarnegar et al. 2016), eCLIP (enhanced CLIP) optimized RNA and cDNA adaptor ligation to improve library generation efficiency and yield (Van Nostrand et al. 2016, 2017b), and other groups have explored orthogonal improvements like utilizing linear amplification via T7 transcription to further reduce amplification bias (Su et al. 2021). Further improvements to these and related protocols have continued to enhance the ability of profiling RBP–RNA interactions from in vivo samples, fewer cells, and at improved scale (Buchbender et al. 2020; Blue et al. 2022; Masuda et al. 2020). The above approaches have dramatically improved our ability to profile the interactome for an individual RBP, enabling profiling at previously intractable scales such as profiling interactions for 150 RBPs (Van Nostrand et al. 2020a,b), and more recently more than 100 zinc-finger proteins (Gosztyla et al. 2024).
To accelerate the comprehensive profiling of the entire landscape of protein–RNA interactions, several innovative methods have been developed that enable the simultaneous profiling of multiple RBP–RNA interactions in a single experiment using barcode-based strategies. For instance, antibody-barcode (ABC) eCLIP conjugates unique DNA barcodes to RBP antibodies followed by proximity ligation to link the barcode to RNAs that interact with that specific RBP (Lorenz et al. 2023). Similarly, SPIDR (split and pool identification of RBP targets) utilizes a split-and-pool barcoding approach to uniquely label individual antibodies and their associated RNA molecules (Wolin et al. 2025). With such antibody-specific barcoding, it is now possible to perform parallel identification of RNA interactomes for multiple to dozens of RBPs simultaneously (Lorenz et al. 2023; Wolin et al. 2025). Further developments in this area are rapidly progressing—for example, PRIM-seq introduces a strategy in which RBPs are cotranslationally “barcoded” by maintaining interaction with their own mRNA sequence, enabling antibody-independent profiling of protein–RNA interactions (Qi et al. 2025).
Crosslinking
To enhance the UV crosslinking efficiency, PAR-CLIP (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation) incorporates a photoactive ribonucleoside, such as 4-thiouridine (4sU), into nascent RNA followed by UVA/B (365 nm) treatment (Hafner et al. 2010). Although this method significantly improves the RNA recovery, its reliance on metabolic labeling and cytotoxicity in certain cell types limits broader application. Alternatively, to increase the crosslinking efficiency of RNAs with proteins that may weakly or indirectly contact RNA, a low concentration of formaldehyde (0.1%) has been used (Hendrickson et al. 2016; Patton et al. 2020; Xiao et al. 2024). Formaldehyde introduces covalent bonds between nearby proteins and nucleic acids, thereby capturing not only direct RNA–protein contacts but also protein–protein (and their associated RNA) interactions. This mild, reversible crosslinking approach can help stabilize low-affinity interactions that may be underrepresented in UV-based methods and help reveal extensive RNA interactions for chromatin regulators as well as regulatory functions of noncoding RNAs in epigenetic control of gene expression. Efforts continue to develop chemical crosslinkers with improved functionality, including recent work indicating that NHS-diazirine can label protein–RNA interaction sites for subsequent sequencing readouts (Weidmann et al. 2021). More exotic approaches, such as genetically incorporating a latent bioreactive unnatural amino acid fluorosulfate-l-tyrosine (FSY) into targeted proteins allows for RBP-specific crosslinking to nucleophilic groups of bound RNAs (Sun et al. 2023), may provide orthogonal opportunities to improve crosslinking in a protein-specific manner.
Methods to enrich for an RBP of interest
As immunoprecipitation of a protein–RNA complex of interest is a key aspect of CLIP, a variety of methods have been adopted to ensure both recovery and specificity of the RBP of interest. Most commonly this relies on high-quality, specific antibodies, and extensive efforts have been made to catalog and validate antibodies that can successfully immunoprecipitate RBPs (Sundararaman et al. 2016; Van Nostrand et al. 2020a). However, many proteins still lack suitable antibodies. Exogenous expression of an RBP open reading frame coupled with epitope tags such as FLAG or V5 provides a solution to this problem, as commercial immunoprecipitation-grade antibodies for these epitope tags enable standard CLIP profiling (Van Nostrand et al. 2017a). To further enhance the specificity of enrichment for the tagged protein–RNA complex, uvCLAP (ultraviolet crosslinking and affinity purification) incorporated tandem His-tag affinity purification, allowing for pure isolation compared to antibody-dependent approaches (Maticzka et al. 2018). GoldCLIP (gel-omitted ligation-dependent CLIP) and SpyCLIP (SpyTag-based CLIP) similarly utilized covalent binding to a ligand followed by high-stringency, denaturing washes to enable purification of the protein of interest using the HaloTag–HaloLink and SpyTag–SpyCatcher systems, respectively, eliminating the need for complex SDS-PAGE electrophoresis and nitrocellulose membrane transfer steps from traditional CLIP protocols (Gu et al. 2018; Zhao et al. 2019; Zhang et al. 2020). For all of these cases, in the case of concern that overexpression of a protein of interest could lead to nonphysiological interactions, these tags can be integrated into endogenous RBP loci via CRISPR/Cas9-mediated homologous repair, followed by standard CLIP approaches (Van Nostrand et al. 2017a).
While all these improvements have substantially expanded our understanding of regulatory functions of RBP–RNA complex, ongoing challenges continue to drive the refinement of these techniques. For instance, numerous RBPs exhibit dynamic subcellular localization, prompting the development of approaches such as Fr-iCLIP (fractionation iCLIP), which integrated CLIP protocols with cellular fractionation to reveal compartment-specific RNA targets of SR proteins (Sanford et al. 2008; Brugiolo et al. 2017). More recently, proximity-labeling strategies, including dibromofluorescein (DBF)–mediated or APEX2-mediated approaches, have been incorporated into CLIP protocols, offering a promising alternative for spatially resolved RBP–RNA interaction profiling (Engel et al. 2021; Yi et al. 2024). In addition, as many RBPs act both individually and as part of multiprotein complexes, methods such as RiPiT (RNA:protein immunoprecipitation in tandem) and Re-CLIP have utilized sequential immunoprecipitation of two different factors to explore subcomplex specificity (Singh et al. 2012; Mabin et al. 2018; Ducoli et al. 2025). These and other efforts have continued to expand our ability to understand RBP-mediated regulatory networks at increasing scale and resolution, providing an enabling technology for the exploration of how RBPs control gene expression and human physiology.
IDENTIFICATION OF PROTEIN-ASSOCIATED RNA–RNA INTERACTIONS
Although many RBP–RNA interactions are guided by specificity derived from RNA binding domains within the RBP, RBP–RNA interactions can also be mediated by guide RNAs that act in RNP complexes, like miRNAs and snoRNAs. With further elucidation of the essential role of such regulatory RNAs in human biology has come significant efforts to adapt RBP target profiling methods to more directly explore these RNA–RNA interactions.
Traditional CLIP of RNPs that incorporate RNA–RNA interactions could identify both the small RNA and target RNAs as separate sequencing reads. As most regulatory RNAs act through complementarity (whether of the entire RNA, or a smaller seed or guide region), it is possible to use such separate mapping approaches to identify enriched target regions, and then predict what guide RNA drove that enrichment through seed matching (Chi et al. 2009; Hafner et al. 2010; Gumienny et al. 2017). To reduce the rate of false-positives, several computational approaches combining CLIP data sets with expression-based data sets were developed to more precisely predict miRNA targets using features such as flanking sequences and target conservation (Wen et al. 2011; Erhard et al. 2013; Majoros et al. 2013; Liu and Wang 2019; Huang et al. 2022a; Uthayopas et al. 2024). As an orthogonal approach, purified AGO protein preloaded with a desired miRNA can be incubated with a presynthesized library of RNA sequences, followed by purification and sequencing of bound RNAs (McGeary et al. 2019). This adaptation of the “Bind-n-Seq” approach (Lambert et al. 2014) allows quantitative and comprehensive measurement of how protein-dependent RNA interactions are altered by seed and extended pairing or mismatches, enabling a deep characterization of the targeting rules for miRNAs (McGeary et al. 2019).
Similarly, enrichment and sequencing of RNA bound to PIWI proteins has provided unique insights into the broader diversity and molecular roles of piRNAs. piRNA biogenesis follows a “ping-pong” cycle where antisense piRNAs are generated and loaded into aubergine (Aub) or PIWI proteins, which cleave the sense strand of transposons; the cleaved fragments are then processed to form sense piRNAs, which recognize and cleave the antisense precursor piRNA in a reciprocal loop that generates a dynamic pool of piRNA sequences (Brennecke et al. 2007; Kim et al. 2009; Senti and Brennecke 2010; Czech and Hannon 2016). Characterization of piRNA interactions via Bind-n-Seq of piRNA-loaded PIWI proteins recently revealed how increased mismatch tolerance for piRNA interaction and cleavage revealed insights into the evolutionary role of piRNAs in transposon silencing (Gainetdinov et al. 2023).
To directly identify RNA–RNA interactions in vivo at transcriptome-wide scale via high-throughput sequencing, chimeric CLIP/CLASH (crosslinking ligation and sequencing of hybrids) was developed (Fig. 1, center). In this approach, protein-bound RNA duplexes are crosslinked, immunoprecipitated, and then directly ligated together to create “chimeric” RNAs (Kudla et al. 2011; Helwak et al. 2013; Helwak and Tollervey 2014; Broughton et al. 2016). Although low efficiency of chimeric ligation remains a challenge that limits recovery of chimeric reads, these chimeric reads represent a unique improvement that enables direct mapping of RNA–RNA interactions. To further improve the protocol, the chimeric ligation strategy was subsequently integrated into various CLIP-based methods, including HITS-CLIP, iCLIP, irCLIP, PAR-CLIP, and eCLIP, giving rise to derivative techniques such as CLEAR-CLIP (Moore et al. 2015), hiCLIP (Sugimoto et al. 2015, 2017), irCLASH (Song et al. 2020), iPAR-CLIP (Grosswendt et al. 2014), and chimeric eCLIP (Manakov et al. 2022; Song et al. 2025). The same chimeric ligation strategy can also be applied in a protein-independent manner to identify RRIs (Fig. 1, right), as utilized by methods such as PARIS (psoralen analysis of RNA interactions and structures) (Lu et al. 2016), LIGR-seq (ligation of interacting RNA followed by high-throughput sequencing) (Sharma et al. 2016), SPLASH (sequencing of psoralen crosslinked, ligated, and selected hybrids) (Aw et al. 2016; Shenasa et al. 2025), and KARR-seq (kethoxal-assisted RNA–RNA interaction sequencing) (Wu et al. 2024) as recently comprehensively reviewed (Ye et al. 2024).
By coupling RRI detection with immunoprecipitation or purification of specific RBP handles, protein-dependent RRI techniques enhance the signal for the specific subset of interactions that involve RNAs of interest. To date, these approaches have successfully identified the interactomes of a variety of small RNAs, including miRNAs via enrichment of AGO2 and other argonaute proteins (Helwak et al. 2013; Kishore et al. 2013; Grosswendt et al. 2014; Helwak and Tollervey 2014; Moore et al. 2015; Kim and Kim 2019; Manakov et al. 2022), snoRNAs utilizing FBL and other snoRNP-associated proteins (Dudnakova et al. 2018; Elliott et al. 2025; Dunn-Davies et al. 2025; Song et al. 2025), and piRNAs via enrichment of Piwi protein PRG-1 (Shen et al. 2018; Ariura et al. 2024), significantly advancing our understanding of the regulatory roles of these noncoding RNAs in gene expression. They have played particularly critical roles in elucidating the targeting rules for the necessary complementarity for miRNA binding (Helwak et al. 2013; Grosswendt et al. 2014; Helwak and Tollervey 2014; Moore et al. 2015; Kim and Kim 2019; Manakov et al. 2022), uncovered novel snoRNA–target interactions (particularly in tRNAs, snRNAs, and other non-rRNA targets) (Dudnakova et al. 2018; Elliott et al. 2025; Dunn-Davies et al. 2025; Song et al. 2025), and helped understand the elusive mechanisms of how piRNAs play particularly important roles in the germline (Shen et al. 2018; Ariura et al. 2024). Moreover, these approaches have been applied to profile double-stranded RNA (dsRNA) binding proteins including Staufen1 (STAU1), ADAR, and DROSHA, uncovering intramolecular interactions which contribute to the structural organization of mRNAs and pri-miRNAs and elucidate how these structures shape gene regulation (Sugimoto et al. 2015; Kim and Kim 2019; Song et al. 2020).
These approaches are highly adaptable to diverse systems, and have been similarly utilized to aid in the characterization of small RNA interactomes in E. coli (Waters et al. 2017; Iosub et al. 2020), Salmonella enterica (Liu et al. 2023), Caenorhabditis elegans (Shen et al. 2018), and Drosophila (Ariura et al. 2024), thereby advancing our understanding of noncoding RNA-mediated regulatory functions across evolution. With the widespread application of these techniques across species and molecular contexts, databases have been established to integrate all these experiments, facilitating access to RRIs (Lee et al. 2025; Li et al. 2025; Yang et al. 2025).
COMPUTATIONAL ANALYSIS OF PROTEIN-ASSOCIATED RRIs
The rapid advancement of experimental methodologies has catalyzed the development of bioinformatic platforms aimed at detecting chimeric RNA reads. Although each approach employs its own computational pipeline for chimeric read identification, the overarching workflow remains largely consistent across methods. This typically includes a number of steps that are common to CLIP and other similar methods, including demultiplexing, adaptor trimming, and removal of PCR duplicates that utilize common tools (e.g., cutadapt [Martin 2011], UMI-tools [Smith et al. 2017]), as well as one key distinction: the identification of chimeric reads via stepwise mapping and hybrid interaction prediction (Fig. 2).
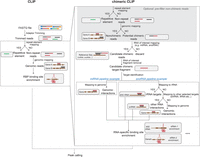
Workflow of computational pipeline in CLIP and chimeric CLIP protocols. (Left) Analysis for CLIP and chimeric CLIP involves standard adaptor trimming, genomic mapping, and peak calling. (Right) Specific modifications for chimeric CLIP include (top) premasking of nonchimeric reads, (middle) an initial mapping pass to identify candidate chimeric reads, and (bottom) single or sequential mapping steps to map the remaining chimeric target fragment with high specificity.
The key question in the identification of chimeric reads is to identify those reads that contain a breakpoint for which the 5′ and 3′ ends clearly map to distinct transcripts. This necessitates mapping reads using “local alignment” modes that do not require the full read to align to a target, which is accommodated by standard alignment tools including Bowtie (Langmead and Salzberg 2012) and STAR (Dobin et al. 2013). However, there is currently no standardized framework for evaluating multialignment results or maximizing chimeric read recovery—each analysis retains sub-read mapping based on criteria such as highest alignment score, customized mismatch scores, and maximum alignment length to exclude low-quality alignments, but achieving an optimal tradeoff between sensitivity and accuracy remains challenging. The incorporation of linker sequences between RNA fragments in hiCLIP and MARIO simplifies this process, facilitating clear separation and reducing ambiguity at the ligation junction (Nguyen et al. 2016; Sugimoto et al. 2017). We also found that a prefilter by performing an “end-to-end” alignment of reads to repetitive elements and the genome, followed by removing these end-to-end aligning reads as nonchimeric reads, could remove likely false positive chimeras before chimera identification (Song et al. 2025).
The complexity of separating hybrid reads, and subsequently the mapping approach, differs based on the RNA types. For unbiased transcriptome-wide detection of RRIs, this typically requires a stepwise approach—reads are first mapped with local alignment, regions that are successfully mapped are removed, the remaining “candidate” chimeric fragment is saved if it is of sufficient size to map (>18–20 nt), and then these are mapped to identify the final paired RNA–RNA interactions (Fig. 2). Although these mapping steps can be performed against the entire genome or transcriptome, as protein-associated RRI mapping often focuses on specific RNA families, the specific details of these RNAs can enable more tailored analysis protocols. For example, rather than performing the initial mapping of potential chimeric reads to the entire genome, restricted mapping can be performed to a curated database of RNAs (e.g., miRNAs, snoRNAs, or piRNAs) to increase sensitivity. Similarly, the second chimeric mapping step can be optimized based on expected targets. For example, most snoRNAs guide interactions with either ribosomal RNA (rRNA) or spliceosomal RNAs (snRNA), each of which has multiple expressed and pseudogene copies in the genome that can cause multi- or mis-mapping. Thus, for snoRNA analysis, mapping putative chimeras sequentially to rRNA, then proceeding with unmapped reads against databases of snRNAs, tRNAs, and only then mapping as-yet unmapped reads to the full genome, can increase the recovery of known interactions and remove false positives (Song et al. 2025).
Strategies for mapping RNAs of interest also vary depending on RNA types. For instance, mature miRNAs are a relatively static size (20–22 nt long) that do not need to be fragmented (and indeed would become too short to map if fragmented), and tend to form a straightforward chimeric ligation product with the miRNA on the 5′ end and the target RNA on the 3′ end (Hafner et al. 2010; Moore et al. 2015). This short static size makes it more effective to align mature miRNA sequences against the reads (Moore et al. 2015; Manakov et al. 2022). In contrast, larger RNAs like snoRNAs and snRNAs (∼100–200 nt), which possess complex second structures, require fragmentation to enable chimeric ligation and sequencing. This leads to unpredictable ligation patterns around base-paired regions, resulting in variably hybrid reads that require mapping against the full snoRNA or snRNA sequence (Song et al. 2025). Similarly, the processing of piRNAs generates a variety of mature small RNAs, requiring reads to first be mapped against either precursor transcripts or the genome (Bagijn et al. 2012; Lee et al. 2012; Zhang et al. 2018).
After computational identification of chimeric reads, validation remains a key question. In most cases, initial validation utilizes sequence properties to ensure that identified chimeric interactions have expected complementarity (Helwak and Tollervey 2014; Dudnakova et al. 2018; Manakov et al. 2022; Dunn-Davies et al. 2025; Song et al. 2025), and in many cases known targets can be used to perform standard sensitivity versus specificity analysis (Grosswendt et al. 2014; Song et al. 2025). Subsequent functional validation depends on the type of RNA under study—candidate microRNA interactions can be validated by overexpression or knockdown of the miRNA of interest followed by RNA-seq or translation profiling (Helwak et al. 2013; Grosswendt et al. 2014; Manakov et al. 2022), whereas validation of snoRNA interactions requires knockdown followed by profiling RNA modification status or target RNA functions (Gumienny et al. 2017; Dudnakova et al. 2018; Song et al. 2025).
UNIQUE EXPERIMENTAL CHALLENGES AND SOLUTIONS FOR IDENTIFYING RBP-ASSOCIATED RRIs
As might be expected, utilizing protein-based RRI mapping approaches involves a number of specific challenges compared to traditional RBP target analysis from CLIP or similar methods. Regardless of the RNA type of interest, protein-centric RRI methods share a similar overall workflow: crosslinking the protein–dsRNA complex, chimeric ligation to join the RNA–RNA interactors, enrichment of desired RNA fragments, protein–RNA hybrid purification, and library preparation steps (including reverse transcription and cDNA amplification) (Figs. 1, 3). With each of these steps comes unique challenges for protein-based RRI mapping, and a variety of adaptations have been developed to address these challenges. Below, we highlight the strengths and challenges associated with each step, providing guidance for researchers seeking to apply these methods.
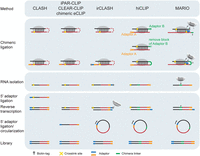
Comparison of chimeric ligation strategies and library preparation in RRI methods. Most methods now incorporate a T4 RNA ligase step without adaptors to encourage direct proximity ligation of base-paired RNAs. To further improve chimeric ligation efficiency and selectively enrich chimeric reads, hiCLIP and MARIO utilized a linker or biotinylated linker to bridge RNA duplexes. The 3′ cDNA adaptor can be added either to linear or circularized cDNA before PCR amplification. Additionally, irCLASH conjugates infrared dye and biotinylated adaptor for RNA visualization.
Crosslinking the protein–dsRNA complex
Regulatory RNA interactions with their targets create a region of dsRNA, which typically acts as the focal point for association of the RNP complex. As such, a key aspect of enrichment for this interacting region involves crosslinking of the associated proteins to (or at least nearby) the dsRNA region. Crosslinking with UVC (254 nm) remains the predominant method in most IP-based methods, including CLASH, hiCLIP, irCLASH, and chimeric eCLIP (Sugimoto et al. 2015; Helwak and Tollervey 2016; Song et al. 2020, 2025; Manakov et al. 2022). However, as base-pairing prevents protein interaction with the nucleoside Watson–Crick face, a protein–dsRNA interaction would not be expected to crosslink with UV. Consistent with this prediction, data from hiCLIP revealed that UV crosslink sites do not typically occur within the RNA duplex regions but rather at sites upstream of the RNA duplex (Sugimoto et al. 2015). A similar observation of crosslink sites predominantly localized to regions upstream of or downstream from RNA duplexes, rather than within the base-paired regions themselves, was observed with 4sU treatment coupled with UVA/B (365 nm) in iPAR-CLIP (Grosswendt et al. 2014). Thus, CLIP profiling of dsRNA-associating RBPs involves the protein components primarily crosslinking to single-stranded regions adjacent to the dsRNA region. To enhance crosslinking efficiency of proteins to dsRNA regions, formaldehyde has been recruited in approaches including fCLIP and MARIO, enabling the profiles of proteins that bind to dsRNA, such as DROSHA and Staufen1 (Ricci et al. 2014; Nguyen et al. 2016; Kim et al. 2017; Kim and Kim 2019).
However, improvement of protein–RRI crosslinking remains an open challenge of great interest to this field. One appealing avenue is to build upon recent success in developing novel chemical crosslinking reagents that capture and profile protein-independent RNA–RNA interactions. Psoralen and its derivate 4′-aminomethyltrioxalen (AMT) can intercalate into RNA duplexes upon UV irradiation at 365 nm, inducing covalent interstrand crosslinks between adjacent pyrimidine bases. These reagents have been utilized in PARIS, SPLASH, and LIGR-seq (Aw et al. 2016; Lu et al. 2016; Sharma et al. 2016; Han et al. 2022), revealing conserved architectures of mRNAs and many novel ncRNA–RNA interactions. SHARC utilizes dipicolinic acid imidazolide to reversibly crosslink 2′-hydroxyl groups of ribose in RNA, enabling the profiling of RNA 3D structures at nanometer-scale (Van Damme et al. 2022). KARR-seq utilized N3-kethoxal followed by dendrimer-mediated crosslinking of proximal RNAs to label RNA with azide groups, enabling the identification of RNA tertiary structures and uncovering intermolecular viral–host RNA interactions and snoRNA–mRNA interactions (Wu et al. 2024; Liu et al. 2025). Although these chemical-based approaches currently are challenging for nonexperts (often requiring specialized synthesis expertise) and in some cases have low efficiency in crosslinking protein-protected RRIs, future optimizations may enable significantly more efficient stabilization and pulldown of protein-mediated RRIs.
RBP–RNA complex precipitation (IP)
The advantages and limits of antibody-based immunoprecipitation for CLIP and relevant techniques have been widely discussed (Wheeler et al. 2018). Like CLIP, this step is also critical for the success of IP-based RRI profiling techniques, yet it faces the same technical challenge inherent to CLIP approaches. Efficient pulldown and proper purification strategies can significantly increase signal-to-noise ratio by removing nonspecific signal. Most methods use antibodies to pull down endogenous or exogenous protein-of-interest; although this often uses standard CLIP immunoprecipitation and SDS-PAGE steps, we recently observed that the SDS-PAGE and nitrocellulose membrane transfer step could be replaced with a simple proteinase K digestion for AGO2 chimeric experiments without loss of specificity (Manakov et al. 2022). However, other methods remain under development—for example, MARIO characterizes the global RRIs bound by all proteins in the cell by biotinylating cysteine residues in all proteins, followed by biotin-streptavidin pulldown to allow for denaturing wash conditions (Nguyen et al. 2016). Stringent wash conditions showed decreased background for microRNA interactions using HIS-tagged AGO2 and would be expected to similarly work for Halo-tag and SPY-tag variants; however, it should be noted that if this denaturing wash is performed prior to chimeric ligation, the yield is substantially decreased (as both small RNA and target would have to independently crosslink to the protein to remain associated) (Helwak et al. 2013; Helwak and Tollervey 2016).
RNA duplex chimeric ligation
The cross-ligation of proximal RNAs into a single “chimeric” fragment is a key molecular step that underpins nearly all efforts to map RNA–RNA interactions. However, this ligation is often inefficient, with typically less than 2% of sequenced reads containing such chimeras (Helwak and Tollervey 2014; Manakov et al. 2022; Song et al. 2025). Thus, both enabling as well as optimizing this reaction has emerged as a critical step for improving the scalability and utility of these approaches.
Fragmentation of RNA into the appropriate size for subsequent library preparation steps is an essential part of CLIP and other protocols, as fragments that are too small will be unable to be mapped to the genome, but fragments that are too large will not PCR amplify or sequence properly (Fan et al. 2015; Van Nostrand et al. 2016). This step is even more critical for RRI approaches, as successful RNA ligation with T4 ligase requires the presence of flexible single-stranded RNA regions and the spatial proximity of the exposed RNA ends (Kaufmann et al. 1974). This is particularly challenging to optimize for large RNA molecules with complex structure, likely contributing to the inability to generate chimeras we observed for H/ACA-box snoRNAs (Song et al. 2025). Indeed, hiCLIP of Staufen1 and chimeric eCLIP of AGO2 both indicated that optimized RNase treatment conditions result in an increased yield of mRNAs or miRNAs chimeric reads, with decreased chimeric yield observed with either too much or too little fragmentation (Sugimoto et al. 2015; Manakov et al. 2022). Therefore, optimization experiments are essential to determine the appropriate RNase treatment necessary for different cell types or tissues, and it is often challenging to determine the optimal balance without a sequencing readout.
Among existing methods, most RRI approaches directly ligate RNA duplexes using T4 RNA ligase. This ligation can occur during the normal RNA adaptor ligation step of CLIP protocols (indeed, multiple efforts utilized standard CLIP profiling of RBPs like AGO2 and FBL coupled with computational identification of chimeric reads to identify RRIs [Wen et al. 2011; Hafner et al. 2012; Gumienny et al. 2017]), but many recent methods add an RNA ligase step without adaptors to further encourage such chimeric ligations (Kudla et al. 2011; Grosswendt et al. 2014; Helwak and Tollervey 2014; Moore et al. 2015; Manakov et al. 2022; Dunn-Davies et al. 2025; Song et al. 2025). hiCLIP uniquely incorporated an additional step before ligation, in which an adaptor is ligated between the intended ligation sites (Sugimoto et al. 2015, 2017). This modification was utilized to enable capture of RNA–RNA interactions that lacked an overhanging single-stranded region following fragmentation, but linker-mediated chimeric reads remain rarer than directly ligated chimeric reads, indicating the need for further optimization of this strategy (Chakrabarti et al. 2023). The MARIO approach introduces a biotinylated linker, enabling the specific enrichment of adaptor-ligated chimeric products (Fig. 3; Nguyen et al. 2016).
The separation of genuine RRIs from experimental artifacts is another major area of focus. Although input controls can be used effectively to remove abundant background in standard CLIP (Van Nostrand et al. 2016), their use is less clear for RRI mapping—interacting RNAs will be present in such inputs, and the presence of chimeric reads in these control samples has been reported (Grosswendt et al. 2014), suggesting that they may reflect real interactions and not artifactual signal. In addition to simple read-depth cutoffs, the use of known interactions as true positives, or orthogonal data (e.g., genes with altered expression upon overexpression or knockdown of a miRNA) can help in the identification of optimal criteria for classifying likely interactions from sequenced chimeric reads.
Additionally, nonspecific post-lysis interactions remain a significant concern in all protein-mediated RRI approaches. In standard CLIP, UV crosslinking ensures that RBP–RNA binding occurs prior to cell lysis; however, the chimeric ligation of interacting RNAs means that only one needs to crosslink to the protein handle, making it possible for post-lysis RNA interactions to create chimeric fragments that persist through stringent washes. Multiple groups have tested the rate of these types of false positives by spiking lysate from different species (e.g., E. coli, yeast, Drosophila, mouse) into human lysate and quantifying the frequency of interspecies chimeric ligations. These methods have shown a range of outcomes—from 2% to 10% (Grosswendt et al. 2014; Helwak and Tollervey 2014; Moore et al. 2015; Manakov et al. 2022). However, our recent work with chimeric eCLIP indicated that the fragmentation for the spike-in sample had a significant impact on the rate of these interspecies chimeras, with higher rates observed when the optimal fragmentation conditions were first independently identified for mouse lysate prior to the spike-in experiment (Manakov et al. 2022). Thus, while it is clear that chimeric CLIP approaches generally recover interactions that can be validated by orthogonal approaches, it should be kept in mind that such post-lysis artifacts are possible and that not every observed chimeric read represents a true in vivo interaction.
Reverse transcription and cDNA library preparation
After standard RNA isolation, reverse transcription into cDNA is another step of critical importance for CLIP-based protocols. A key insight in the iCLIP protocol was that proteinase K digestion following UV crosslinking leaves a small amino acid adduct on the crosslinked nucleotide, which often causes traditional reverse transcription reactions to terminate when they reach this nucleotide (König et al. 2010; Huppertz et al. 2014). In traditional CLIP, this termination would lead to the loss of 5′ RNA adaptors, but the truncated cDNA products enable single-nucleotide mapping of crosslink sites if the second adaptor ligation is performed after reverse transcription (Moore et al. 2014; Gillen et al. 2016). For mapping RNA–RNA interactions, however, this represents a more significant challenge—if reverse transcription terminates before reading through both parts of the chimeric RNA, then the interaction information will be lost. To improve the likelihood of reverse transcription readthrough, we utilized modified reverse transcription protocols with Mn2+ buffer that were first developed to improve readthrough of chemical adducts in RNA structure probing experiments (Mortimer et al. 2012; Homan et al. 2014; Siegfried et al. 2014; Smola et al. 2015). We observed these conditions to also read through crosslinked nucleotides in eCLIP (Van Nostrand et al. 2017b; Manakov et al. 2022; Song et al. 2025), allowing the maintenance of as many chimeric reads as possible. The further optimization of reverse transcription conditions should continue to improve the recovery and readout of chimeric reads.
LIMITATIONS AND FUTURE PERSPECTIVES
Despite significant progress in protein-centric RRI mapping, current approaches remain constrained by both technical and analytical limitations. One persistent challenge is the low crosslinking efficiency between RBPs and RNA duplexes. Additionally, all existing techniques suffer from low chimeric ligation efficiency, with only a maximum 2% of sequencing reads representing usable RNA hybrids. Thus, these methods are generally robust for detecting abundant RNA hybrids, such snoRNA–rRNA and miRNA–mRNA interactions, but identifying low-abundance interactions (e.g., snoRNA interactions with rarer snRNAs) remains challenging. Recent efforts, including strategies to deplete abundant RNAs like rRNA (Nguyen et al. 2016; Liu et al. 2025) or to enhance specific RNA–RNA interactions with antisense oligonucleotide probe capture (Manakov et al. 2022), have improved the detection of low-abundant interactions and uncovered novel functional roles of noncoding RNAs. Nevertheless, improving chimeric ligation remains an open challenge (particularly for RNA duplexes embedded within complex structures, like those present at H/ACA box snoRNA:target interactions), underscoring the need for better strategies to enhance ligation efficiency.
From a computational perspective, accurately identifying true RRIs from sequencing data remains a major challenge, with methods to filter high-confidence chimeric reads remaining individually defined across methods and types of small RNA. While approaches such as complementarity prediction have been employed to characterize high-confidence interactions, numerous studies have reported noncanonical miRNA binding sites where 3′ compensatory pairing and centered pairing beyond the seed region make imperfect seed matches sufficient for AGO binding and gene silencing (Grimson et al. 2007; Friedman et al. 2009; Loeb et al. 2012; Seok et al. 2016). This poses a challenge for identifying the full landscape of true miRNA targets with prediction algorithms that depend primarily on seed-matching criteria. The integration of preexisting molecular and biophysical modeling of RNA interactions of interest has provided one solution: Candidate interactions identified from CLIP and chimeric CLIP approaches can be further evaluated by calculating a binding energy for miRNA–target interactions that accounts not only for seed sequence interactions but also for flanking structural features, such as loops, base pairs, bulges, or dangling ends, as well as features of the AGO protein itself, to enable more accurate prediction of miRNA–target interactions (Wee et al. 2012; Khorshid et al. 2013; Breda et al. 2015). In addition, high-throughput functional assays combined with RRI profiling approaches have been utilized to train deep learning models, capable of predicting both seed and seedless sites through integration of the entire miRNA and potential target genes (Pla et al. 2018; Min et al. 2022; Uthayopas et al. 2024). Similarly, focused analysis of snoRNA chimeric reads utilizing a generalized linear model incorporating standard snoRNA:target prediction scoring along with additional sequence and structural features improved recovery of true interactions (Gumienny et al. 2017), and recent work uncovering the interaction dynamics of piRNAs will enable more effective analysis of chimeric piRNA CLIP data (Gainetdinov et al. 2023). These and future efforts to build robust strategies for accurately reconstructing RNA duplex structures from chimeric reads will further aid in determining RRIs at single-nucleotide resolution and help isolate biologically relevant interactions.
The ever-increasing association of regulatory RNAs with disease underscores the need for scalable, accurate methods to directly identify their interactions transcriptome-wide across the variety of tissues and cell types in which they play critical physiological roles. This ongoing methodological innovation in both experimental design and computational analysis to improve the sensitivity, specificity, and interpretability of protein-mediated RRI mapping has provided and will continue to provide unique opportunities to better explore the landscape of how RNA–RNA interactions play critical roles in biology.
COMPETING INTEREST STATEMENT
E.L.V.N. is a cofounder, on the Scientific Advisory Board, equity holder, and paid consultant for Eclipse BioInnovations, on the SAB of RNAConnect, and is an inventor of intellectual property owned by the University of California San Diego. E.L.V.N.’s interests have been reviewed and approved by the Baylor College of Medicine in accordance with its conflict-of-interest policies. The other author declares no competing interests.
ACKNOWLEDGMENTS
We thank members of the Van Nostrand laboratory for their support, insight, and feedback. E.L.V.N. is a CPRIT Scholar in Cancer Research (RR200040) and is supported by the National Human Genome Research Institute (NHGRI, R35HG011909).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080830.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








