
Regulation of cardiac hypertrophy by RNA readers
- Center for Translational and Experimental Cardiology (CTEC), Department of Cardiology, University Hospital Zurich and University of Zürich, Schlieren 8952, Switzerland
- University Heart Center, Cardiology, University Hospital Zurich, Zurich 8091, Switzerland
- Corresponding author: francesco.paneni{at}uzh.ch
A deep understanding of the mechanisms regulating cardiac response to stress is of paramount importance to develop new mechanism-based therapeutic approaches tackling pathological cardiac remodeling in heart failure (Martin et al. 2023). Available therapies exert a significant, yet modest effect on cardiac hypertrophy regression in patients with heart failure with reduced (HFrEF) and preserved ejection fraction (HFpEF) (Martin et al. 2023). This helps to explain the unacceptably high residual risk in heart failure patients despite the availability of effective drugs such as renin-angiotensin-system (RAS) blockers, SGLT2 inhibitors, and GLP1-receptor agonists (Fatima et al. 2023).
Although our genetic makeup is certainly linked to pathological cardiac remodeling (Morita et al. 2008), most of the observed phenotypic variation is driven by nongenetic factors (Costantino et al. 2018). Exposure to environmental stimuli has been shown to derail gene expression during the course of life mainly via chemical modifications of DNA/histone complexes and RNA (Costantino et al. 2018; Wang et al. 2023). Chemical editing of RNA is emerging as a pivotal regulator of several biological processes, such as the stability and translation of mRNA transcripts (Zaccara et al. 2019). Among these modifications, recent work has shed light on methylation of the exocyclic N6-amino group of adenosine (m6A) in mRNA as a pivotal player in the heart (Wu et al. 2020). Yet, our comprehension of m6A signaling and its role in the regulation of cardiac function remains scarce. m6A occurs in RNA molecules near stop codons, in 5′- and 3′-untranslated regions, in long internal exons, and in the shared sequence RRACH (R = G/A and H = A/C/U) (Wu et al. 2020). The m6A modification of RNA is the result of an active interplay among writers (i.e., METTL3), erasers (i.e., ALKBH5 and FTO), and readers (members of the YTH domain family [YTHDF]) (Zaccara et al. 2019).
Mounting evidence indicates that RNA readers (YTHDF family of proteins) are implicated in a wide range of cellular processes including proliferation and metabolism (Chen et al. 2023). YTHDF1 and YTHDF2 were recently shown to be involved in cardiac homeostasis (Golubeva et al. 2023; Gilbert et al. 2024), while the role of YTHDF3 remains poorly understood. Specifically, it remains unclear whether YTHDF3 is redundant or plays an active role in the heart.
In this issue of the journal, Rabolli et al. (2025) unveil a unique function for the RNA reader protein YTHDF3 in the heart (Fig. 1). Bioinformatic analyses of available single-cell datasets in mice and human failing hearts showed a preferential downregulation of YTHDF3 in cardiomyocytes as compared to other cardiac cells, suggesting that this enzyme may be operative in this cell type in conditions of cardiac stress. To determine the in vivo role of YTHDF3 specifically in cardiomyocytes, the laboratory generated a conditional mouse model lacking YTHDF3 in these cells (Y3-cKO). Cardiomyocyte deletion of YTHDF3 was not associated with cardiac phenotypic changes at baseline. Of interest, they observed that Y3-cKO mice had attenuated pathological remodeling following pressure overload injury by transverse aortic constriction (TAC). Specifically, Y3-cKO mice were protected against cardiac hypertrophy, left ventricular dysfunction, and lung congestion. At the cellular level, Y3-cKO mice had preserved cardiomyocyte cross-sectional area, suggesting a potential role of YTHDF3 in regulating cardiomyocyte hypertrophy. Reduced hypertrophy in Y3-cKO mice was also associated with less cardiac fibrosis and inflammation, the latter assessed by interleukin 6 (IL-6) and CXC motif chemokine ligand 10 (Cxcl10). In vitro experiments in rat neonatal cardiomyocytes confirmed the anti-hypertrophic action of YTHDF3 depletion (Rabolli et al. 2025). The authors next performed mass spectrometry–based proteomic analysis to investigate whether YTHDF3 gene silencing is associated with a reshaped cardiomyocyte proteome. Notably, cells lacking YTHDF3 showed a profound alteration of the proteomic landscape with most changes being observed for proteins involved in the regulation of gene expression and metabolism. To test the involvement of YTHDF3 in regulating mRNA translation, a puromycin incorporation assay was performed in control and YTHDF3-depleted cardiomyocytes treated with the growth stimulator phenylephrine. This analysis revealed a lower level of global protein synthesis in the absence of YTHDF3, thus supporting the involvement of YTHDF3 in mRNA translation. Taken together, these data indicate that YTHDF3 is a master regulator of protein synthesis in cardiomyocytes exposed to pro-hypertrophic stimuli. These results were confirmed in vivo, where phenylephrine (PE) stimulation led to hypertrophic growth of cardiomyocytes in control but not in Y3-cKO mice. In these mice, PE-induced protein synthesis was attenuated, thus confirming the central role of YTHDF3 in hypertrophic growth in vivo.
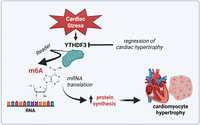
Schematic showing the emerging role of the RNA reader YTHDF3 in cardiomyocyte hypertrophy.
The study by Rabolli et al. (2025) provides new information on the role of the RNA reader YTHDF3 in the heart and pinpoints a novel therapeutic target to prevent hypertrophy in conditions of cardiac stress. Translating these data into patients with heart failure would enable delineation of a molecular target and a potential biomarker of cardiac hypertrophy in this setting. It would also be interesting to investigate whether YTHDF3 deregulation is a common biological signature across different forms of cardiac hypertrophy such as hypertensive cardiomyopathy, aortic stenosis, hypertrophic cardiomyopathy, and other relevant cardiomyopathies where hypertrophic growth is a main feature and an important determinant of cardiac remodeling and mortality. In this perspective, editing YTHDF3 could reduce protein synthesis, thus attenuating hypertrophic growth, diastolic dysfunction, and lung congestion in these conditions.
Besides pro-hypertrophic stimuli, it would also be of high interest to determine whether different forms of cardiac stress, namely metabolic stress (i.e., fatty acids) or toxic stress (cancer-related therapies), result in impaired YTHDF3 signaling and enhanced protein synthesis in cardiomyocytes. YTHDF3 signaling is heavily involved in cancer development (Chen et al. 2025), suggesting that this enzyme could be a critical player at the crossroad of multiple comorbidities and its deregulation could help understanding alterations of cardiac remodeling upon different conditions.
In the present study, YTHDF3 expression was found to be decreased in the cardiomyocyte clusters from mice and humans with heart failure as compared with controls (Rabolli et al. 2025). The downregulation of YTHDF3 in the failing heart could represent a compensatory, yet futile mechanism to overcome the hypertrophic growth and cardiomyocyte damage. Further validation studies are needed to test the biological significance of YTHDF3 downregulation in the failing heart.
Another interesting observation from this study is that YTHDF3 deletion in cardiomyocytes was associated with reduced expression of IL-6. Given the central role of IL-6 in the pathogenesis of heart failure (both HFrEF and HFpEF) and the ongoing efforts to tackle this inflammatory cytokine in this setting (Ridker and Rane 2021; Alogna et al. 2023), the findings by Rabolli et al. (2025) shed light on a new mechanism linking cardiac stress with epitranscriptomic regulation of inflammation in the failing heart. Therapeutic modulation of YTHDF3 could indeed contribute to attenuating hypertrophic growth while blunting cardiac inflammation and fibrosis. The impact of YTHDF3 on fibrosis and inflammation could also be relevant in the setting of ischemic cardiomyopathy where postinfarction remodeling is strongly accompanied by extracellular matrix remodeling and myocardial inflammation (Matter et al. 2024).
Moreover, the role of YTHDF3 in regulating mRNA translation and protein synthesis could heavily influence other biological processes critically involved in cardiomyocyte damage such as defective autophagy and endoplasmic reticulum stress, deregulation of the ubiquitination system, metabolic changes, and pro-apoptotic signals (Lindqvist et al. 2018). Proteomic analyses in YTHDF3-depleted cardiomyocytes showed altered levels of proteins involved in these biological processes (Rabolli et al. 2025).
Additional work is needed to clarify the impact of YTHDF3 on cardiomyocyte metabolism and autophagy and the potential role of YTHDF3-editing strategies in this direction. In conclusion, Rabolli et al. (2025) present an elegant work unveiling a new player involved in cardiomyocyte hypertrophy and a potential therapeutic target in heart failure. Testing YTHDF3-targeting approaches in preclinical models of pressure overload–induced hypertrophy and heart failure could pave the way for new personalized approaches modulating RNA readers in this setting.
ACKNOWLEDGMENTS
We are supported by the Swiss National Science Foundation (no. 310030_197557), the Swiss Heart Foundation (no. FF19045), the Novartis Foundation for Medical and Biological Research, the Foundation for Scientific Research at the University of Zurich, the Olga Mayenfisch Foundation, the Swiss Life Foundation, the Kurt und Senta-Hermann Stiftung, the EMDO Stiftung, and the Schweizerische Diabetes-Stiftung (to F.P.); the Holcim Foundation and the Swiss Heart Foundation (to S.C.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080557.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








