
A general and biomedical perspective of viral quasispecies
- Esteban Domingo1,
- Brenda Martínez-González2,3,
- Pilar Somovilla1,4,
- Carlos García-Crespo1,
- María Eugenia Soria1,3,
- Ana Isabel de Ávila1,
- Ignacio Gadea3,5 and
- Celia Perales2,3
- 1Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), 28049 Madrid, Spain
- 2Department of Molecular and Cell Biology, Centro Nacional de Biotecnología (CNB-CSIC), Consejo Superior de Investigaciones Científicas (CSIC), 28049 Madrid, Spain
- 3Department of Clinical Microbiology, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital, Universidad Autónoma de Madrid (IIS-FJD, UAM), 28040 Madrid, Spain
- 4Departamento de Biología Molecular, Universidad Autónoma de Madrid, 28049 Madrid, Spain
- 5Centre for Biomedical Network Research on Infectious Diseases (CIBERINFEC), 28029 Madrid, Spain
- Corresponding authors: edomingo{at}cbm.csic.es, celia.perales{at}cnb.csic.es
Abstract
Viral quasispecies refers to the complex and dynamic mutant distributions (also termed mutant spectra, clouds, or swarms) that arise as a result of high error rates during RNA genome replication. The mutant spectrum of individual RNA virus populations is modified by continuous generation of variant genomes, competition and interactions among them, environmental influences, bottleneck events, and bloc transmission of viral particles. Quasispecies dynamics provides a new perspective on how viruses adapt, evolve, and cause disease, and sheds light on strategies to combat them. Molecular flexibility, together with ample opportunity of mutant cloud traffic in our global world, are key ingredients of viral disease emergences, as exemplified by the recent COVID-19 pandemic. In the present article, we present a brief overview of the molecular basis of mutant swarm formation and dynamics, and how the latter relates to viral disease and epidemic spread. We outline future challenges derived of the highly diverse cellular world in which viruses are necessarily installed.
Keywords
ORIGINS AND SCOPE OF THE QUASISPECIES CONCEPT
Quasispecies theory was developed by M. Eigen and P. Schuster in the 1970s in the course of theoretical investigations to explain self-organization of matter in the origin of life; the theory was a productive blend of notions derived from Darwinian evolution and information theory (Eigen 1971; Eigen and Schuster 1979). Its core concepts are: (i) the regular production of error copies during replication of primitive macromolecules (RNA or RNA-like) that carry inheritable information, and (ii) the delimitation of the error rate compatible with maintenance of that genetic information, to avoid its irreversible loss by crossing an error threshold (Eigen and Schuster 1979; Swetina and Schuster 1982). Both concepts have influenced virology for two main reasons: (i) because RNA viruses display high error rates and produce mutant distributions as predicted by quasispecies theory, thus providing adaptability to their populations, and (ii) because virus extinction by excess mutations (akin to the transition into error catastrophe of quasispecies theory) is the basis of lethal mutagenesis, as one of the mechanisms of activity displayed by several currently licensed antiviral agents (Loeb et al. 1999; Crotty et al. 2001; Eigen 2002; Perales et al. 2019; García-Crespo et al. 2024). In addition to influencing virology, quasispecies theory has been amply applied to theoretical and experimental investigations, including replicator networks, dynamics of microbial and tumor cell populations, or structural heterogeneities in prion protein assemblies, among others (the scope of quasispecies for subcellular and cellular systems was reviewed in different chapters of Domingo and Schuster 2016). The impact of the theory has gone well beyond its initial purpose of satisfying one of the requirements for the self-organization of macromolecules in the origin of life (Eigen 1992).
QUASISPECIES THEORY HAND-IN-HAND WITH EXPERIMENTS
A mutual influence between experimental results and theoretical concepts has been permanent since the initial formulation of quasispecies theory. The first connections can be traced back to the work of the groups of S. Spiegelman and C. Weissmann with the Escherichia coli RNA bacteriophage Qβ. Spiegelman's group showed that extensive in vitro replication of Qβ RNA by purified Qβ RNA replicase led to the selection of Qβ RNA error copies that were optimized for rapid replication in the in vitro reaction mixture, to the detriment of their infectivity for E. coli cells (Mills et al. 1967; Levisohn and Spiegelman 1969). This observation on RNA selection, together with theoretical concepts on the coding of information and fitness (replicative capacity) optimization (Hamming 1980; Mandelbrot 1983) were major components of quasispecies theory (Eigen and Schuster 1979; Eigen 1992). C.K. Biebricher and colleagues quantified in vitro Qβ RNA replication kinetics and mutant selection parameters (Biebricher et al. 1985; Biebricher 2008), thus strengthening the connections between theory and experiment in the early stages of quasispecies articulation.
Work of C. Weissmann and colleagues in the 1970s with infectious bacteriophage Qβ was carried out in parallel with the formulation of quasispecies theory, and filled aspects that were complementary to the in vitro RNA replication work. They prepared the first protocol of site-directed mutagenesis to generate Qβ RNA with mutations at preselected sites of its genome. This was a scientific breakthrough that represented the onset of reverse genetics in biology and biomedicine (Weissmann et al. 1977). The site-directed mutagenesis procedure allowed the in vitro synthesis of an infectious specific Qβ point mutant, and the calculation of its rate of reversion to the wild type nucleotide in 10−4 mutations per round of copying (Batschelet et al. 1976; Domingo et al. 1976). The study provided evidence of rapid development of genetic heterogeneity in Qβ populations, and established that fitness of individual biological clones retrieved from a population was lower than the fitness of the complete population (Domingo et al. 1976, 1978). These results on the calculation of an error rate, and the ranking of fitness values of individual genomes within a viral population, mirrored important ingredients of quasispecies theory.
The work with bacteriophage Qβ was followed by investigations on population heterogeneity, mutation-selection, and variant competition dynamics with vesicular stomatitis virus (VSV) and foot-and-mouth disease virus (FMDV) by the teams of J. J. Holland and E. Domingo. Their experiments confirmed the conclusions of the Qβ work, and established links between quasispecies behavior and some postulates and principles of population genetics, with high error rates as a distinctive feature in the interpretation of results. As an outcome of these and other studies, viruses are now perceived as suitable systems to interrogate evolutionary events occurring within much shorter time frames than those required using cellular organisms. In addition, it is now recognized that rapid virus evolution has a decisive impact on viral disease mechanisms, and on disease control strategies, as explained in coming sections. (As reviews on how quasispecies-related research has unfolded during the last four decades, see Holland et al. 1982, Eigen and Biebricher 1988, Novella 2003, Domingo et al. 2021b, Sardanyés et al. 2024.)
MOLECULAR BASIS OF HIGH MUTATION RATES AND MUTANT SPECTRA: INDETERMINATIONS IN VIRUS BEHAVIOR
RNA viruses (and DNA viruses whose replication is catalyzed by low-fidelity DNA polymerases) display mutation rates which are in the range of 10−3 to 10−6 mutations per nucleotide copied (Batschelet et al. 1976; Drake and Holland 1999; Sanjuán et al. 2010). Mutation rates are not invariant; dissimilarities within the above range of values have been recorded with different viruses, mutants of a given virus, or different host cells. Polymerase copying fidelity (sometimes denoted as 1-mutation rate) depends on the structural arrangement of amino acids at (or connected with) the catalytic polymerase domain, which interacts with template and primer RNAs during nucleotide incorporation. Since amino acid substitutions may occur at fidelity-relevant sites in the polymerase or in proteins of the replication complex, the mutation rate is an evolvable trait (Earl and Deem 2004; Wei et al. 2022). Even for a given polymerase complex structure acting on identical template–primer sequences, studies with various polymerases suggest that mutation rates may be contingent upon physiological and metabolic responses that alter nucleotide substrate concentrations, ionic strength, or ion composition at or around the catalytic domain (Arnold et al. 1999; Kumar et al. 2011; Wang et al. 2021a; Pourfarjam et al. 2022; Sabariegos et al. 2022).
The regular production of variant genomes (as a result of mutation, recombination, and genome segment reassortment in the case of segmented genomes), inherent to the replication process, and in some cases with the additional participation of cellular editing activities (that may contribute to mutation and biased hypermutation) has been amply documented with animal, plant, and bacterial RNA viruses. RNA viruses (and also some DNA viruses) are pushed by these various molecular mechanisms to generate complex mixtures of coexisting variants that are dynamic in two senses: (i) because their composition can be perturbed by intrahost and interhost bottleneck events (Pfeiffer and Kirkegaard 2006; Bull et al. 2011), and by the bloc transmission of groups of particles (Kerviel et al. 2021); and (ii) because the incessant introduction of new variants precludes reaching a population equilibrium. In this permanent population disequilibrium, mutant spectrum composition is modified even when replication takes place in the absence of externally applied selective pressures (Moreno et al. 2017; Gallego et al. 2020; Delgado et al. 2021).
The commonly determined consensus (or average) sequence—the one that generally enters sequence data banks—is a weighted average of myriads of genomic sequences; it may not be even identical to a subset of the genomes present in the population (Eigen 2013; Domingo and Perales 2019). This is a critical departure from the classical association of a viral isolate with a genomic nucleotide sequence. A viral isolate is in reality a largely undetermined mutant cloud (Fig. 1A). Individual genomes within the cloud need not have the same biological attributes than the population ensemble or other genomes from the same cloud. When considering in an individual isolate subgenomic RNA stretches (i.e., those encoding the polymerase or any nonstructural or structural proteins, or regulatory regions) rather than the entire genome, the stretch may be dominated by a repeated nucleotide sequence, accompanied of a tail of low-frequency minority mutants. Dominance of a repeated sequence does not apply to the entire viral genome, given the average mutation rates and frequencies calculated for RNA viruses (Batschelet et al. 1976; Domingo et al. 1978; Drake and Holland 1999; Sanjuán et al. 2010). The following numerical example illustrates this point. In a population of a viral genome 10,000 nt in length, we consider a random distribution, with an average of five mutations per genome. Assuming no fitness effects of mutations, the percentage of genomes without mutations predicted by the Poisson distribution is 0.67%. The same calculation for a subgenomic RNA stretch of 3000, 300, and 30 nt (maintaining the same average of five mutations per genome) yields 22.3%, 86.1%, and 98.5%, of genomic stretches without mutations, respectively (Fig. 1A). Even if these percentages are the same with any population size of the genome (or subgenomic stretch) distribution, the actual occupation of sequence space is not. The mutation frequency is independent of the population size (intrinsic property), while the probability of the presence of a specific mutant is higher the larger is the virus population size (extrinsic property) (Domingo and Perales 2012). Adaptability is influenced by the degree of genetic heterogeneity and the virus population size. This is why persistently infected, immunocompromised individuals in whom virus replication is only partially restrained by the immune response (viral “super-shedder” individuals) can produce a large viral load with the potential of increasing the probability of generation of specific mutants that can produce a phenotypic change. Indeed, “super-shedders” have proven an important source of variant viruses with potentially altered traits (Rocha et al. 1991; Dunn et al. 2015; Truong et al. 2021; Igari et al. 2024); poor immune status also correlates with the high diversity of human immunodeficiency virus type 1 (HIV-1) reservoirs in T-cell subsets (Zhang et al. 2024b). The combinatorial possibilities that can potentially result in trait novelty are further underlined by considering that the mutant swarm representation in Figure 1A refers to a virus isolate. Different isolates generally vary in their consensus sequence (Domingo 2020), including epidemiologically closely related SARS-CoV-2 isolates (Delgado et al. 2024). The possible phenotypic effects of mutations may differ by virtue of their being anchored in different sequence contexts.
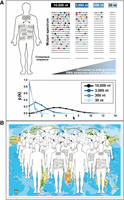
Intrahost viral quasispecies, and their impact in epidemic and pandemic spread. (A) Schematic representation of mutant spectra. On the upper left, an infected individual is viewed as including multitudes of compartmentalized mutant distributions; in reality, they are of far larger population sizes than drawn in the picture for simplicity. On the right, genomes or subgenomic stretches are drawn as horizontal lines, and mutations are depicted as symbols on the lines. Assuming an average of five mutations per a 10,000 nt genome, the number of genomes without mutations is minimal; the number increases when shorter, subgenomic RNA stretches (length in nucleotides [nt] indicated in the upper boxes) are considered (lines not drawn to scale). The graphs below indicate the probability of occurrence of viral genomes with k mutations [p(k)] per genome or subgenomic RNA stretch (color code for RNA length in the inserted box), according to the Poisson distribution, assuming an average of five mutations per genome. A numerical example and the importance of the virus population size for exploration of the space of functionally relevant sequences are explained in the text. (B) Representation of the pandemic spread of a virus from infected to susceptible individuals. Mutant distributions, captured in population bottlenecks of different intensity (arrows), are the transmitted entities. The complexity of mutant spectra adds to the uncertainties inherent to modifications of virus behavior following expansions among susceptible individuals. For simplicity, in A and B we display only point mutations, but for some RNA viruses (particularly SARS-CoV-2), insertions, and more often deletions, also contribute to genomic and subgenomic RNA variations.
Positive and negative epistasis—profusely described in viruses (Sanjuán et al. 2004; Elena et al. 2010; Innocenti et al. 2024; Schwab and Yin 2024)—is an additional influence that affects virus traits by mutations in distant genes of the same genomic molecule or mutations at different positions within the same gene. Given the proportion of genomes with two or more mutations in mutant distributions (Fig. 1A) and that the viral population size in infected individuals (even immunocompetent) can reach up to 1012 infectious units (for review, see Domingo 2020), there is ample opportunity for some mutations pairs or clusters to exert epistatic effects. Traits resulting from positive epistasis may be expressed or remain suppressed by the complete population. Therefore, time-dependent population heterogeneity and viral population size, in conjunction with influences among mutations within the same genome, or interactions among genomes, creates indeterminations in virus behavior. Uncertainties are further intensified by differences among individual hosts and within a host. Genetic (often also epigenetic) differences, with metabolic consequences, are now recognized as a widespread feature of unicellular and multicellular collectivities, including cell-to-cell variations within the same tissue (Murante and Hogan 2024; Schaible et al. 2024; Swaminath and Russell 2024).
Accounts of unexpected behavior of viruses are found in the literature, particularly in medical virology. The following statements by J. J. Holland and colleagues are relevant to the matter: “Because a particular RNA virus simply does not exist, a particular RNA virus disease does not exist either.… These facts are easily overlooked by clinicians and scientists because disease syndromes are often grossly similar for each type of virus, and because it would appear to make no difference in a particular sense. However, for the person who develops Guillain–Barré syndrome following a common cold, or for the individual who remains healthy despite many years of HIV-1 infection, it may make all the difference in the world.” (Holland et al. 1992) (Guillain–Barré syndrome is a muscle weakness condition caused by damage of the peripheral nervous system. Damage is mediated by an autoimmune disorder that may be triggered by a viral infection.) Indeterminations have been evidenced once again with COVID-19 by remarkable variations in disease severity and sequelae even within each pandemic wave (Vihta et al. 2022).
Low-fidelity replication, the molecular event that underlies quasispecies formation, has been extensively supported by measurements of incorporation of correct versus incorrect nucleotide substrates, using purified RNA-dependent RNA polymerases (RdRps) and RNA-dependent DNA polymerases (RdDps; reverse transcriptases) in a variety of in vitro nucleotide incorporation assays. These enzymes often lack proofreading-repair (3′-5′ exonuclease) activities that could decrease nucleotide misincorporation rates. Such an absence (or low efficiency) is in contrast to their presence in the DNA-dependent DNA polymerases (DdDps) that catalyze replication of complex DNA viruses or cellular DNA. In addition, the nascent DNA products of such replicative DdDps benefit of postreplicative repair pathways that can further reduce the mutational input (Gartner and Engebrecht 2022; Herbst et al. 2024). Despite being infrequent, mechanisms of error correction are not totally alien to RNA replication machineries. They have been described for influenza virus (IV), hepatitis C virus (HCV), HIV-1, and satellite RNAs of some plant viruses (for review, see Domingo et al. 2021a). Their presence may modulate but not suppress the production of variant genomes.
Some coronaviruses express a 3′-5′ exonuclease (Exo N) that reduces the error rate of the polymerase by about 15-fold, as calculated for the coronavirus murine hepatitis virus (Eckerle et al. 2007). An active Exo N is also encoded in the SARS-CoV-2 genome, but its contribution to lowering the error rate during RNA genome replication is unknown. In vitro measurements with the SARS-CoV-2 holo-polymerase complex (nsp12-nsp8-nsp7, without the nsp14 that includes the Exo N) have yielded error rates of 10−1 to 10−3 misincorporations per nucleotide (Yin et al. 2023). This range implies a 10- to 2000-fold higher introduction of erroneous nucleotides than that determined with in vitro assays for other RdRps, such as those of picornaviruses (Arias et al. 2008; Cameron et al. 2009). Mutation rates of around 10−6 mutations introduced per nucleotide copied during SARS-CoV-2 replication in infected cells have been indirectly estimated from mutation frequencies (Amicone et al. 2022; Kawasaki et al. 2023). Independently of the accuracy of such measurements, the available data indicate that this virus fully participates in the implications of quasispecies dynamics, despite its large genome size and the encoded Exo N (Andrés et al. 2020; Karamitros et al. 2020; Armero et al. 2021; Butler et al. 2021; Lythgoe et al. 2021; Tonkin-Hill et al. 2021; Bader et al. 2022; Dudouet et al. 2022; Kimbrel et al. 2022; Martínez-González et al. 2022a,b; Karakasiliotis et al. 2023; Delgado et al. 2024). Not only reports on SARS-CoV-2 population heterogeneity, but also those on variation in disease symptoms and transmissibility, selection of antiviral-escape mutants and vaccine failures, further support the embedding of this pathogen in quasispecies implications (Wang et al. 2021b; Hachmann et al. 2022a,b; Tuekprakhon et al. 2022; Hirotsu et al. 2023; Ameratunga et al. 2024; Tanino et al. 2024; Zhang et al. 2024a). The picture with SARS-CoV-2 regarding population complexity and its medical connotations is similar to that exhibited by other pathogenic RNA viruses (for review, see Domingo et al. 2012, 2023).
EXPERIMENTAL OBSERVATIONS AND THEORETICAL ADJUSTMENTS
Mutual influences between new experimental results and accommodation of quasispecies theory to those results, have contributed to interpret the adaptability of virus populations, and to plan control disease strategies accordingly. Developments worth underlining have included: (i) extensions of quasispecies theory to finite populations in variable fitness landscapes (Saakian and Hu 2016; Schuster 2016; Schuster and Stadler 2023); (ii) inclusion of recombination in the quasispecies formulations (Boerlijst et al. 1996; Jacobi and Nordahl 2006); (iii) demonstration of a molecular memory in viral quasispecies, as a reflection of their behaving as complex adaptive systems. It is manifested by an increase in the frequency of some mutations in a viral population, as the result of fitness gain that the genomes harboring such mutations undergo in the process of their selection from the mutant spectrum. Since mutation frequency depends on genome fitness, mutations in memory genomes are found at a higher frequency than they would have been in the absence of the process of selection. This type of memory equips viral populations with a mechanism of rapid adaptation to environmental changes that were previously experienced by the same evolutionary virus lineage, provided population bottlenecks did not intervene (Ruiz-Jarabo et al. 2000; Briones and Domingo 2008); (iv) evidence of limited tolerance of viruses to error rates that exceed their basal levels (Holland et al. 1990). This led to lethal mutagenesis as an antiviral strategy, akin to the transition into error catastrophe through violation of the error threshold relationship of quasispecies theory (Loeb et al. 1999; Eigen 2002; Perales et al. 2019); (v) recognition of intrapopulation complementation and cooperation (Shirogane et al. 2016), as mechanisms that contribute to quasispecies behaving as a unit of selection; (vi) new information on population composition drawn from the application of ultradeep sequencing methodologies, which allow capturing the magnitude of intrapopulation complexity and dynamics of viral populations. The sequence space occupied by viruses, and the possibilities of transitions between different points of sequence space, are far more extensive than initially anticipated (Sardanyés et al. 2024).
Although not a comfortable viewpoint change, the current picture of RNA virus populations transmits the need to confront a transition from the focus on “sequences” to a focus on “genome clouds.” This would help to interpret virus behavior more accurately, and to redefine the concept of residue conservation, with all its biotechnological implications. Indeed, at least for HCV, the number of conserved residues is significantly higher when consensus instead of mutant spectrum sequences of the same populations are aligned (García-Crespo et al. 2020). This difference in conservation criterion is relevant as a demonstration of the capacity of mutant spectra to explore regions of sequence space that are not represented by consensus sequences. The difference is also highly pertinent to the design of universal vaccines or pangenomic antiviral agents, which is often based on the less strict conservation criteria derived from the alignment of consensus sequences in data banks (García-Crespo et al. 2020).
NEW DIMENSIONS OF COMPLEXITY: RECOMBINATION AND DEFECTIVE GENOMES
Quasispecies theory, which was initially centered on point mutations, was extended to incorporate recombination and its influence on fitness landscapes and the position of the error threshold for maintenance of genetic information (Boerlijst et al. 1996; Jacobi and Nordahl 2006; Saakian 2018). Intramolecular and intermolecular recombination occurs with high frequency in viruses whose replication is catalyzed by low processivity polymerases or polymerase complexes (those that tend to detach from the template molecule they are using to synthesize the complementary strand). The polymerase may jump from one template to another, or from one site to another site of the same template molecule. The result is the generation of mosaic genomes of standard length and genomes with insertions and deletions. Probably, some sites have sequence features that favor polymerase detachment and template rebinding; that is, recombinant break points are not random (Wang et al. 2022). However, detailed studies with enteroviruses have indicated that many incipient recombinant constructs are eliminated by negative selection due to their low replicative fitness (Bentley et al. 2021; Alnaji et al. 2022). Only part of the recombination events that actually take place can be identified through marker mapping and phylogeny of the progeny genomes. Recombination has probably contributed to the origin of new viruses or part of their genome complement: The coronavirus mouse hepatitis virus (MHV) may have captured its hemagglutinin-esterase gene from a type C IV (Luytjes et al. 1988); the alphavirus Western equine encephalitis virus probably arose by recombination between a virus related to Eastern equine encephalitis virus and another similar to Sindbis virus (Weaver 2006). Thus, recombination is a driving force of short-term and long-term evolution of some RNA viruses.
Insertion and deletion mutants, together with missense mutations, contribute to the pool of defective genomes (DGs). The DG frequency varies widely among viruses, depending on the recombination frequency dictated by the replication machinery, type of host cell, capacity of trans-complementation of defective gene products, and evolutionary history (distance of the population from a bottleneck founder event, and occurrence of massive infections or bloc transmission of multiple particles). In the recently emerged SARS-CoV-2, infectious genomes coexist with abundant DG subsets characterized by in-frame and out-of-frame deletions (which lead to premature termination codons), or other lethal point mutations. The latter are not easy to distinguish from mutations that while not being lethal, lead to strong reduction of viral fitness. The result is the coexistence of two classes of genomes other than the standard, infectious genomes (which display a relative fitness close to the maximum): bona fide DGs (that require complementation for their maintenance), and low fitness (albeit viable genomes) (Campos et al. 2022; Hillung et al. 2024). The presence of an abundant subset of low fitness genomes agrees with the abundance of low-frequency mutations (in the 0.1%–0.5% frequency range quantified with current ultradeep sequencing procedures) and haplotypes detected in SARS-CoV-2 nasopharyngeal isolates (Martínez-González et al. 2022a; Delgado et al. 2024). The absence of intermediate frequency mutations in SARS-CoV-2 mutant spectra marks a difference with HCV quasispecies from chronically infected patients, as examined with comparable ultradeep sequencing methodology (Martínez-González et al. 2022a).
The presence in the same replicating population of genomes with different length has been recapitulated with the “ultracube” concept (Sardanyés et al. 2024), as an extension of the hypercube, used to depict movements in sequence space (Eigen and Schuster 1979; Swetina and Schuster 1982). Vertices of a hypercube correspond to positions in the sequence space of genomes with the standard length (without insertions or deletions), with edges representing mutations that connect vertices. In the ultracube extension, several interconnected hypercubes coexist, each of them corresponding to a genome length category. Movements in sequence space consist not only in point mutations that connect vertices of a hypercube, but also movements among hypercubes mediated by insertions and deletions. Ultracubes conceptualize the complexity of viral populations that exhibit high recombination rates (Sardanyés et al. 2024). All mechanisms of genome modification (point mutations, recombination, segment reassortment in segmented genomes) are compatible modes of variation that contribute to short-distance and long-distance evolutionary changes.
MEDICAL IMPLICATIONS OF ENHANCED COMPLEXITY DUE TO THE DG POOL
DGs are reservoirs of mutated viral RNA regions that can be reintroduced into infectious genomes by recombination. Because of this, DGs can contribute to the therapeutic implications of quasispecies dynamics—in particular to the selection of antiviral-resistant mutants—that were previously centered on infectious genomes (Ribeiro et al. 1998; Perales et al. 2012, 2020; Wang 2024). This participation may be particularly relevant for SARS-CoV-2, since viable genomes and an ample DG pool coexist in natural isolates and laboratory populations, in a recombination-prone genetic system (Jackson et al. 2021; Sekizuka et al. 2022; Fang et al. 2023; Karim et al. 2024; Sayama et al. 2024). The understanding of SARS-CoV-2 DG structure and interfering activity learned from DG purification and interference assays is still largely pendent.
DGs vary in structure and in the intensity and mechanism by which they interfere with the infectious cycle of the corresponding standard genomes (Giachetti and Holland 1989; Finke and Conzelmann 1999; Pelz et al. 2021; Brennan and Sun 2024). Unless DGs are generated at very high rates and remain only temporarily in the population (with a survival time dictated by the half-life of the RNA molecules), their continued presence requires trans-complementation of defective functions, supplied by expression products of the standard genome.
Inhibitory DG clouds promote the evolution of their standard genome counterparts; this was established in the case of defective interfering (DI) particles, initially of VSV by observing the competition-selection dynamics of alternating mutant forms of DIs and standard genomes (Palma and Huang 1974; Semler and Holland 1979). Depending on their frequency in the viral RNA pool and their inhibitory capacity, DGs may mediate the establishment of persistent infections or modulate immune responses, in addition to being reservoirs of variant sequences (Huang and Baltimore 1970; Holland and Villarreal 1974; Vignuzzi and López 2019; Brennan and Sun 2024; González Aparicio and López 2024; Hillung et al. 2024).
Flow of RNA between defective and infectious genomes by recombination implies that the DG pool can play a role in two major medical implications of quasispecies: (i) As a determinant of viral pathogenesis, manifested in the adaptive invasion of tissues within an organism. This process can be favored by mutations whose consequences can be as diverse as an expansion of host cell tropism or the production of a wider mutant spectrum (Baranowski et al. 2001; Pfeiffer and Kirkegaard 2005; Vignuzzi et al. 2006, among other examples). (ii) As contributors to failures in prevention (vaccine inefficacy, selection of pathogenic revertants of live attenuated vaccines), and therapeutics (selection of antiviral-escape mutants). Recombination involving infectious or DGs has been shown to be a source of multidrug-resistant virus variants (Kellam and Larder 1995; Moutouh et al. 1996; Fraser 2005; Kouyos et al. 2009; Quan et al. 2009; Mukherjee et al. 2011). This mechanism of multidrug escape is particularly adept in HIV-1 where the recombination rate exceeds the mutation rate by threefold (Mansky and Temin 1995; Jetzt et al. 2000).
Strategies to circumvent these quasispecies-related medical problems have met with partial success. Except for disease associated with antigenically constant viruses (typically measles, rabies, and hepatitis A virus as human pathogens), the majority of RNA viral diseases require complex, multiepitopic vaccines to achieve a reasonable level of protection. Furthermore, vaccine efficacy necessitates periodic updating of its antigenic composition, to match the antigenic profile of the circulating virus. Comparable requirements apply to antiviral agents. The latter should be administered in combination because monotherapy is synonymous with viral breakthrough (selection of drug-escape mutants) and treatment failure, as profusely documented with many viral pathogens (Ribeiro et al. 1998; Perales et al. 2012, 2020; Wang 2024). Similarly, monoclonal antibody monotherapy is an invitation to transient benefits, and to the selection of antigenic variants of viral pathogens. Successful antiviral therapies (i.e., HIV-1 suppression, or HCV sustained virological response and virus clearance) are often achieved with combination therapies, but with the problem of selection of treatment-escape mutants not entirely eliminated (Wang 2024). Despite quasispecies-driven prevention and treatment failures, and means to prevent them having been suggested decades ago (Domingo 1989; Domingo and Holland 1992), antiviral and antibody monotherapies are still sometimes practiced for reasons that are not clear to us.
In this scenario of viral disease control difficulties, lethal mutagenesis has irrupted as a broad-spectrum antiviral design that stems from the error threshold concept of quasispecies theory (Eigen 2002; Perales et al. 2019). Despite all lethal mutagens (base and nucleoside analogs or their pro-drugs, which are intracellularly converted into their active nucleoside-triphosphate forms) targeting primarily the viral polymerase, they can act synergistically, as recently documented with SARS-CoV-2 (Abdelnabi et al. 2021; García-Crespo et al. 2024). In this and other studies (for review, see Perales et al. 2019), the transition toward virus extinction ran parallel with elevation of viral mutation frequency. Two features of the nucleotide analogs encourage the exploration of synergistic lethal mutagenesis as a treatment for emergent RNA virus infections: (i) They often display other mechanisms of antiviral activity in addition to lethal mutagenesis, and (ii) the preferred mutation types and genome sites where such mutations occur may differ among analogs (Gallego et al. 2019). These two attributes favor synergism, according to pharmacology tenets. In cell culture model systems, synergistic lethal mutagenesis was effective to extinguish HCV—a virus whose polymerase is not known to encode an exonuclease repair activity—(Gallego et al. 2019), and SARS-CoV-2—a virus whose polymerase encodes an ExoN—(García-Crespo et al. 2024).
FACTS AND INFERENCES ON THE QUASISPECIES CONTRIBUTION TO VIRAL DISEASE EMERGENCE AND PANDEMIC SPREAD
Genome adaptability is one among diverse factors (sociological, political, environmental, climatic, technological) that have been identified as potentially playing a role in the emergence of transmissible diseases (Morse 1994; Smolinski et al. 2003; Morens and Fauci 2020). The process of viral disease emergence has been divided in three steps: introduction, establishment, and dissemination. Introduction refers to the initial event by which a virus present in a given biological species (either causing pathology or not) infects an individual of a different host species and causes disease. Establishment indicates the multiplication of the introduced virus not only in the first individual, but also in its immediate contacts, implying a capacity of host-to-host transmission in the new host species. Dissemination designates the overt spread of the virus among many individuals of the new host species, to give rise to disease outbreaks, epidemics, or pandemics.
Viral disease emergence has been documented in several biological groups, particularly animals and plants, and it is assumed that there are abundant introduction events that do not progress toward the establishment step. Viruses can be transmitted from animals to humans (zoonotic origin of a new human virus), as in the case of IV or SARS-CoV-2 (Woo et al. 2006; Li et al. 2019; Holmes 2024) and vice versa (reverse zoonosis); for example, swine vesicular disease virus most likely originated from human coxsackievirus B5 (Graves 1973; Zhang et al. 1999; Bruhn et al. 2015). Phylogenetic evidence suggests frequent interspecies transmissions of parvoviruses (Hoelzer and Parrish 2008), and circulation of SARS-CoV-2 between humans and animals (Rasmussen et al. 2024), among other observations on the transit of viruses among species. A widely accepted view is that viral particles—that constitute the virosphere (fortunately with a great majority of nonpathogenic viruses), and that are 10 times more abundant than cells in our biosphere (Koonin and Dolja 2013)—are in continuous flow among cellular organisms, particularly in the present, highly interconnected world. Viral traffic largely depends on biological transmission vectors (notably arthropods, birds, mammals) that often modify their habitability zones and migratory routes due to climate change and agricultural and commercial practices. Wet-markets were recognized as a source of human respiratory infections well before the emergence of SARS-CoV-2 (Woo et al. 2006). Both, the stochastic nature of genetic modifications and the many changing influences involved in virus traffic, render viral emergences the unpredictable outcome of a vast lottery.
The question we address in the context of the present article is how quasispecies dynamics can impact any of the three steps in viral disease emergence, in an attempt to distinguish facts from indirect inferences. To document a direct role of quasispecies in the introduction step of a virus in a new host species (be animal or plant), is not within the capabilities of current surveillance practices. Such documentation would require witnessing the moment at which a virus enters productively for the first time an individual of a new host species, and then to identify the genomes from the mutant spectrum that entered the new host, using ultradeep sequencing of virus samples from the donor and recipient individual. Even if such surveillance-experiment were feasible, it would be very difficult to show that a subset of genomes (and not a different subset or any subset) from the cloud of the donor individual was necessary for the initial replication of the virus in the new host. A requirement of a virus being a mutant cloud—rather than a defined genomic sequence—for the initial introduction to be successful is presently an unverifiable event. There is no direct evidence (if fact, there cannot be with the available methodology) that quasispecies per se is a necessary determinant of the introduction step in the emergence of a viral disease.
There is, however, extensive evidence that minority components of viral populations exhibit features that differ from those of the dominant genomes, and that render the virus fit to enter a new host species. Viral populations include minority variants that display altered host cell tropism, which is a relevant trait both, for viral disease emergence and viral pathogenesis, two interconnected concepts. Among several other examples (for review, see Domingo 2020), substitutions in the capsid protein were identified by clonal analyses of the apathogenic prototype strain of the parvovirus minute virus of mice during its infection of severe combined immunodeficient (SCID) mice. Recombinant viruses harboring the relevant mutations exhibited lower avidity than wild type virus for permissive fibroblast cell lines, and caused lethal disease following oronasal inoculation of SCID mice. The single substitutions influenced pathogenicity and adaptation to a new host (López-Bueno et al. 2006).
Substitutions in the viral capsid were also involved in an expansion of cell receptor recognition of coxsackievirus B (CVB). This virus group uses a receptor termed the coxsackievirus and adenovirus receptor (CAR) to enter cells (Bergelson 2010). Upon passage of CVB in a cell line that expressed a limited amount of CAR, a small number of capsid amino acid substitutions were selected that expanded the cell receptor specificity to allow the virus to bind CAR and also the alternative receptor decay accelerating factor (DAF) (Carson et al. 2011).
The identity or physical overlap between residues of a viral capsid or envelope that are involved in antibody response and cell tropism promotes coevolution of the two traits (Stewart and Nemerow 1997; Baranowski et al. 2003; Domingo 2020). FMDV antigenic variants were produced upon passage of the virus in cell culture in the absence of antibodies. Some of these variants included substitutions in a protruding Arg-Gly-Asp triplet in the viral capsid that affected both reactivity with antibodies and recognition of the integrin receptor (Verdaguer et al. 1995; Martínez et al. 1997). The Arg-Gly-Asp triplet that previously had been considered essential for integrin receptor recognition and virus viability, turned out not to be necessary for the maintenance of virus infectivity. In absence of the triplet, the virus found alternative pathways for cell entry (Baranowski et al. 2000). Thus, quasispecies evolution in a cell culture environment—unperturbed by external selective pressures—was capable of rendering dispensable a receptor recognition motif that was invariant among natural isolates of the virus. The FMDV antigenic variants achieved the remarkable capacity to infect at least five additional cell lines that were resistant to the parental (unpassaged) virus (Ruiz-Jarabo et al. 2004). The prevalence of antigenic variants in viral populations—as judged from their frequency of 10−3 to 10−5 monoclonal antibody escape mutants in many viral populations (Domingo 2020)—further argues in favor of the presence of cell tropism mutants in viral populations. The continuous confrontation of viruses with the host immune response may have as an indirect (but selectively favored) consequence the exploration of alternative pathways to enter cells.
Nonstructural proteins may be also involved in cell tropism and host range modifications. A substitution in the nonstructural protein 3A was critical for the adaptation of a clonal population of a swine FMDV to the guinea pig (Núñez et al. 2001). Remarkably, mutations that were adaptive for the new guinea pig host did not preclude maintaining the capacity to productively infect the original swine host (Núñez et al. 2007). In this case, quasispecies memory (Ruiz-Jarabo et al. 2000; Briones and Domingo 2008) may have played a role in such rapid readaptation. Through quasispecies memory or by other mechanisms, a capacity to retain the infectivity for the previous host species is advantageous for the virus and may have contributed to several human-to-animal transits of SARS-CoV-2 (Rasmussen et al. 2024). Quasispecies favored the cyclic replication of the dual-tropic measles virus in lymphatic tissue and in epithelial cells prior to transmission (Donohue et al. 2019). To what extent quasispecies is a more general means to safeguard reversibility of cellular and host preferences is worth exploring.
Several epidemiological and experimental observations support a role of quasispecies in the establishment and dissemination steps of viral disease emergence. The efficacy of adaptation of the morbillivirus canine distemper virus to ferrets varied depending on the mutant spectrum complexity of the canine virus. A recombinant that exhibited lower population diversity than an attenuated (Vero cell-adapted) counterpart led to delayed onset of disease symptoms in the ferret (Siering et al. 2024); the nonrecombinant virus gradually increased the frequency of beneficial mutations that preexisted in the mutant spectrum.
Basic theoretical considerations and results from experimental evolution converge to conclude that viruses allowed to replicate without population restrictions in a given environment will increase their fitness in that environment. Fitness gain will proceed until population size limitations preclude further increase (Novella et al. 1995a,b, 1999; Escarmís et al. 1999; Jerzak et al. 2005; Nijhuis et al. 2009). A relationship between quasispecies composition and epidemiological dissemination has been drawn from combining virological and bioinformatics approaches. The intrahost population diversity that resulted from a higher than average polymerase error rate of some norovirus lineages enhanced the virus pandemic potential (Bull et al. 2010). Algorithms have been developed to infer transmission virus routes from the intrahost quasispecies composition, with applications to HIV-1 and HCV (Glebova et al. 2017). SARS-CoV-2 mutant spectrum composition can be informative of transmission routes and of genomic sequences which are likely to become dominant at later stages of the pandemic spread of the virus (Choi et al. 2020; Sun et al. 2021; Martínez-González et al. 2022b,c, 2024; Messali et al. 2023). SARS-CoV-2 mutant spectra may explore new constellations of mutations and deletions that may feed future genomes that contribute to sustain the dissemination step. Virus transmission among individuals, particularly transmission of respiratory viruses, usually entails some population bottleneck (Fig. 1B). In contrast, different transmission ways that have been documented with other viruses involve the absence of (or less severe) bottlenecks. For example, massive viral transmissions occurred with HCV and HIV-1 when blood transfusions were performed at the time when diagnostic reagents for the presence of these two viruses in donors' blood were not available. During their expansion among susceptible individuals, RNA viruses evolve at rates that are in the range of 10−2 to 10−5 mutations introduced per nucleotide and year, with values that depend on several factors, including the time elapsed between the sampling of viruses whose sequences are compared (specific examples and discussion of evolutionary rates in Domingo 2020).
The syllogism: virus adaptability is important for viral disease emergence, quasispecies is important for adaptability, therefore quasispecies is important for viral disease emergence, may be regarded as too simple. However, several lines of evidence just outlined (notably the continuous exploration of sequence space, the abundance of cell tropism mutants and the ease for viruses to find alternative cell entry pathways, coevolution of antigenicity and cell tropism, faster adaptability to a new host of heterogeneous viral populations than their more homogeneous counterparts, mutant spectra as factories of epidemiologically relevant viruses) strengthen the connections between quasispecies dynamics and part of the requirements for a viral infection to emerge and to be sustained in a new host species. Further studies are needed to elucidate in molecular detail how specific components of mutant spectra can contribute to a virus emergence. The challenge is remarkable due to indeterminations in virus population composition and behavior.
CONCLUSION AND OUTLOOK
The present article has summarized the origin of the quasispecies concept, and the influence it has exerted on virology (Table 1). The central point of such influence was the recognition that RNA virus populations are dynamic mutant clouds of an undetermined composition that nevertheless guides their adaptability to changing environments. The clouds do not act as mere independent mutant aggregates since interactions among variants of the same cloud can influence behavior. Dynamic complexity and unpredictability are accentuated by the absence of population equilibrium in mutant spectra since their composition is altered by the stochastic generation of genomic changes in continuous interaction with a diverse array of host cells and organisms. Mutation (and in some cases also recombination and segment reassortment) is systematic and not occasional. These molecular mechanisms push viral populations toward diversification, and one of the recent surprises has been that SARS-CoV-2, despite its large genome size and an encoded Exo N activity, fully participates in the dynamics that results in vaccine failures and selection of antiviral-resistant mutants. To what extent the ExoN activity attenuates these manifestations of quasispecies adaptability is an open question. Adaptability also translates into the need to apply multiple selective constraints to effectively suppress viral replication. Despite difficulties in demonstrating a direct participation of the quasispecies nature of viruses in viral disease emergence, there is reasonable evidence that intrapopulation diversity and the presence of certain variant types may have facilitated some of the steps in viral disease emergence.
Main points on viral quasispeciesa
There are many pendent questions open to theoretical and experimental research to further penetrate into the adaptive mechanisms displayed by viruses, and how to improve medical interventions. Not alien to the challenge is the diversity of the cellular word. In fact, the discovery of viral quasispecies in the presequencing, pre-PCR era preceded the recognition of biological complexity of the cellular world. Intrapopulation differences among individual cells have been substantiated by genomic surveys and single-cell analyses. Concepts on biological complexity and how to deal with it are shared by viruses, cancer cell populations, microbial collectivities, and even prions, where conformation heterogeneity and state transitions take the place of genetic change in viruses.
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Ciencia e Innovación, grants PID2020-113888RB-I00/AEI/10.13039/501100011033 and 202220I116, the Ministerio de Ciencia, Innovación y Universidades (MICIU), grant PID2023-146622OB-I00 financed by MICIU/AEI/10.13039/501100011033, and by FEDER, UE, through the “Severo Ochoa” Programmes for Centres of Excellence in R&D (CEX2021-001154-S and CEX2023-001386-S). The work was also supported by the European Commission-Next Generation EU (regulation EU 2020/2024) through the CSIC's Global Health Platform (PTI Salud Global). The work was also funded by grants PI21/00139 funding from Instituto de Salud Carlos III (ISCIII), cofunded by the European Union, CSIC-COV19-014 from Agencia Estatal Consejo Superior de Investigaciones Científicas (CSIC), and project 525/C/2021 from Fundació La Marató de TV3, grants 202136-30 and 202136-31. We also acknowledge the project S2018/BAA-4370 (PLATESA2 from Comunidad de Madrid/FEDER). Institutional grants from the Fundación Ramón Areces and Banco Santander to the CBMSO are also acknowledged. The team at CBMSO belongs to the Global Virus Network (GVN), and to the Biomedicine Unit of Universidad de Castilla La Mancha (UCLM), associated with CSIC. B.M.-G. is supported by predoctoral contract PFIS FI19/00119 from Instituto de Salud Carlos III cofinanced by Fondo Social Europeo (FSE), “El FSE invierte en tu futuro.” P.S. is supported by postdoctoral contract Margarita Salas, CA1/RSUE/2021 from MCIU. C.G.-C. was supported by predoctoral contract PRE2018-083422 from MCIU.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080280.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








