
RNA sensing at the crossroads of autoimmunity and autoinflammation
- 1National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), National Institutes of Health (NIH), Bethesda, Maryland 20892, USA
- 2RNA Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, USA
- Corresponding authors: sandra.williams{at}nih.gov, sandra.wolin{at}nih.gov
Abstract
Immune-mediated diseases are common in humans. The immune system is a complex host defense system that evolved to protect us from pathogens, but also plays an important role in homeostatic processes, removing dead or senescent cells, and participating in tumor surveillance. The human immune system has two arms: the older innate immune system and the newer adaptive immune system. Sensing of foreign RNA is critical to the innate immune system's ability to recognize pathogens, especially viral infections. However, RNA sensors are also strongly implicated in autoimmune and autoinflammatory diseases, highlighting the importance of balancing pathogen recognition with tolerance to host RNAs that can resemble their viral counterparts. We describe how RNA sensors bind their ligands, how this binding is coupled to upregulation of type I interferon–stimulated genes, and the ways in which mutations in RNA sensors and genes that play important roles in RNA homeostasis have been linked to autoimmune and autoinflammatory diseases.
Keywords
- RNA sensors
- Toll-like receptors
- RIG-I-like receptors
- type I interferons
- autoimmune disease
- autoinflammatory disease
INTRODUCTION
The immune system evolved to protect the host from pathogens. Pathogens must first be recognized as foreign. However, recognition of pathogens must be balanced with tolerance to host molecules, as breaks in tolerance lead to autoimmunity—the immune system aberrantly recognizing the host as foreign (Pisetsky 2023). Pathogen recognition activates different signaling cascades that are meant to clear the pathogen and limit its ability to damage the host (Mogensen 2009). In mammals, there are two branches of the immune system. The newer branch of the immune system is the adaptive immune system (Chi et al. 2024). The effector cells of the adaptive immune system are T and B lymphocytes, and these cells recognize specific molecular patterns through T and B cell receptors, which undergo extensive genetic rearrangements to allow them to “adapt” to the many specific pathogens that may be encountered.
The older branch is the innate immune system, which serves as the first line of defense against pathogens and acts rapidly. The effector cells of this system are primarily myeloid cells. These cells phagocytose pathogens, release antimicrobial and cytotoxic granules that are directly toxic to pathogens, activate and recruit additional immune cells by secreting cytokines and chemokines, and activate the adaptive immune system through antigen presentation (Carpenter and O'Neill 2024).
Pattern recognition receptors (PRRs) are critical components of the innate immune system. These proteins detect an array of pathogen-associated molecular patterns (PAMPs) and damage/danger-associated molecular patterns (DAMPs). PRRs evolved to recognize exogenous glycans, lipids, DNA, and RNA. They are found in cellular compartments that are sites of pathogen entry, such as the cell surface, endosome, and cytosol. Ligand binding to a PRR activates a host defense signaling cascade that results in an antimicrobial and pro-inflammatory response.
This Perspective will focus on RNA sensors, which are a class of PRRs that primarily function in antiviral responses. While enveloped viruses can directly fuse with the plasma membrane to enter the cell, viruses can also be endocytosed (Fig. 1). Viral uncoating can occur in the endolysosomal compartment or in the cytosol, which exposes the nucleic acid contents of the virus (Rybicki 2023). This is necessary for replication but also allows viral nucleic acid to be recognized by PRRs. Whether RNA is detected in the endosome or in the cytosol, RNA binding to PRRs results in signaling through adapter proteins to induce transcriptional activation of inflammatory cytokines and type I interferon–stimulated genes (ISGs; Fig. 1). These ISGs play broad roles in the antiviral response.
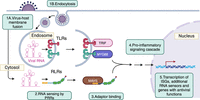
Viral RNA is sensed by pattern recognition receptors and results in transcriptional activation of type I interferon–stimulated genes (ISGs) and an antiviral response. Viruses enter cells by fusing with the plasma membrane or by endocytosis. After uncoating, viral RNA is exposed to the cell. Toll-like receptors recognize RNA in the endosome, and RIG-I-like receptors recognize RNA in the cytosol. They signal through adapter proteins to activate a pro-inflammatory signaling cascade and transcription of ISGs, including additional RNA sensors and genes that function in the antiviral response (e.g., IFITs, RNase L, PKR). (Image was created in BioRender.com.)
While RNA sensors evolved as part of the host's antiviral defense system, aberrant RNA sensing and type I interferon signaling are implicated in some autoimmune and autoinflammatory diseases, including systemic lupus erythematosus (SLE) and Aicardi-Goutières syndrome (AGS). In these diseases, the aberrant activation of these pathways significantly contributes to the pathogenesis of these diseases.
TOLL-LIKE RECEPTORS AND RIG-I-LIKE RECEPTORS ARE THE PRINCIPAL ANTIVIRAL RNA SENSORS
The main classes of PRRs that recognize RNA are Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs) (Fig. 1). TLRs are type I transmembrane glycoproteins with a leucine-rich repeat (LRR) domain and a Toll/IL1 receptor (TIR) domain. The LRR domain is the site of ligand binding, and the TIR domain binds to adapter proteins that are essential for propagating the pro-inflammatory signaling cascade. The TLRs that recognize RNA (TLR3, TLR7, and TLR8) are localized to endosomes, with their ligand-binding domains inside the endosome lumen. They are differentially expressed in immune cells and have distinct ligand-binding specificities. TLR3 is expressed in dendritic cells (Muzio et al. 2000) and B cells, as well as fibroblasts, epithelial cells, and endothelial cells. TLR3 recognizes long (>40 bp) double-stranded RNAs (Tatematsu et al. 2013), which are produced during replication of many viruses (Weber et al. 2006). TLR3 dimers can assemble into a higher order multimeric complex along dsRNA, which enhances signal transduction (Sakaniwa et al. 2023). TLR3 signals through the adapter molecule TRIF (TIR-domain-containing adapter-inducing interferon-β) (Fig. 2A; Oshiumi et al. 2003). This ultimately leads to activation of IRF3/IRF7, as well as NF-κB (Meylan et al. 2004), resulting in transcription of inflammatory cytokines and type I ISGs.
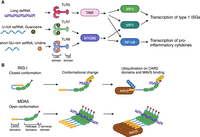
Toll-like receptors and RIG-I-like receptors recognize different features of RNAs. (A) Toll-like receptors. TLR3 recognizes long double-stranded RNA and signals through the adapter TRIF. TLR7 recognizes single-stranded GU-rich RNA and binds guanosine at a second site. TLR8 recognizes short single-stranded GU-rich RNA and binds uridine at a second site. Both TLR7 and TLR8 signal through the cytosolic adapter MyD88. (B) RIG-I-like receptors sense dsRNA. RIG-I binds double-stranded sequences of at least 10 bp through its helicase domain. The C-terminal domain recognizes the RNA 5′-triphosphate moiety. RNA binding is coupled with derepression of the CARD signaling domains and CARD domain ubiquitination. The CARD domains bind to the adapter protein MAVS. MDA5 binds long double-stranded RNA and assembles into helical filaments along the RNA through its helicase and C-terminal domains. The CARD domains signal through MAVS. (Image was created in BioRender.com.)
TLR7 is expressed predominantly in plasmacytoid dendritic cells, and to a lesser extent in B cells and monocytes/macrophages. It recognizes single-stranded U-rich RNA (Zhang et al. 2016, 2018), as well as guanosine at a second binding site, and signals through the MyD88 adapter protein (Fig. 2A), leading to activation of IRF7 and NF-κB and transcription of type I ISGs and inflammatory cytokines. Interestingly, 2′,3′-cGMP has a higher affinity than guanosine for the guanosine binding site, suggesting increased sensitivity to RNA degradation products (Zhang et al. 2018). TLR8 is expressed in myeloid cells and recognizes short single-stranded GU-rich RNA that may be generated by RNA degradation (Tanji et al. 2015; Ostendorf et al. 2020), as well as uridine at a second site (the dimerization interface) (Tanji et al. 2015). Like TLR7, it signals through MyD88 (Fig. 2A). The preference of TLR7 and TLR8 for single-stranded U- or GU-rich RNA, respectively, may allow for recognition of broad classes of viral RNAs, while the preference of both TLRs for degradation products may reflect the fact that the endolysosomal compartment contains abundant nucleases. Indeed, RNase T2 and other nucleases generate ligands from microbial RNAs that activate TLR7 and TLR8 (Greulich et al. 2019; Ostendorf et al. 2020; Berouti et al. 2024; Nunes et al. 2024). Host RNAs escape recognition, since their cytosolic location precludes recognition by endosomal TLRs. In addition, some uridines in host RNAs are modified to pseudouridine (Carlile et al. 2014; Schwartz et al. 2014), which decreases their immunogenicity and prevents recognition by TLRs (Kariko et al. 2005).
In contrast to the TLRs, whose ligand-binding domains are within endosomes and are confined to specific cells of the immune system, RLRs are cytosolic and expressed in most cell types (Fig. 2B). Both MDA-5 and RIG-I contain tandem signaling domains called caspase recruitment domains (CARDs) at the N terminus, a central DExD/H box helicase domain, and a C-terminal domain. LGP2, another RLR, lacks the N-terminal CARDs and thus does not have an independent role in signal transduction. RIG-I is the best studied of these proteins, and in the absence of a ligand, is in an autorepressed, closed conformation with the CARDs sequestered through interactions with the helicase domain (Kowalinski et al. 2011). Ligand binding results in a large conformational change that releases the CARDs, allowing them to become competent for downstream signaling. Upon derepression, the CARDs undergo ubiquitination and bind the adapter protein MAVS (Gack et al. 2007; Oshiumi et al. 2010; Kuniyoshi et al. 2014). RIG-I prefers to bind double-stranded sequences of at least 10 bp with a 5′-triphosphate moiety (Linehan et al. 2018; Ren et al. 2019). The C-terminal domain is important for 5′-triphosphate recognition (Cui et al. 2008; Ren et al. 2019). The 5′ ends of mRNAs are modified by the addition of a 7-methylguanosine cap and by ribose 2′-O-methylation of nucleoside(s) immediately adjacent to the cap, which prevents these RNAs from activating RIG-I (Schuberth-Wagner et al. 2015; Devarkar et al. 2016; Durbin et al. 2016). For RNA polymerase III transcripts, the 5′ end is often removed during maturation or sequestered by proteins.
While MDA5 has a very similar domain architecture, the CARDs exist in a more open conformation and are not sequestered in the absence of ligand (Berke and Modis 2012). MDA5 binds long double-stranded RNA (Ren et al. 2019), and MDA5 molecules assemble into helical filaments along the length of the RNA (Fig. 2B; Berke and Modis 2012; Yu et al. 2018). In contrast to RIG-I, the C-terminal domain does not bind the RNA triphosphate. It does, however, participate in filament-forming interfaces (Yu et al. 2018). The helicase and C-terminal domain of MDA5 form a flexible dsRNA-binding interface that can accommodate different helical twists of dsRNA (Yu et al. 2018). The different MDA5 binding modes are also coupled with distinct occupancy and nucleotide-binding preferences at the ATP-binding site, suggesting an important role for ATP hydrolysis in MDA5 filament formation and signaling (Yu et al. 2018). As with RIG-I, the MDA5 CARDs interact with the MAVS adapter protein to promote transcription of ISGs.
LGP2 binds dsRNA with high affinity but is less discriminating than either RIG-I or MDA5 in its ligands. It binds both short and long dsRNA and binds more tightly to dsRNA than either RIG-I or MDA5 (Saito et al. 2007; Uchikawa et al. 2016). It does not require a 5′-triphosphate (Uchikawa et al. 2016). While it is not as well understood as the other RLRs, it has a complex array of regulatory roles and has both positive and negative regulatory effects on RIG-I and MDA5 (Bruns and Horvath 2015; Rehwinkel and Gack 2020).
ADDITIONAL RNA SENSORS ARE UPREGULATED UPON PRR ACTIVATION
As described above, a common consequence of activation of the RNA-sensing TLRs and RLRs is the transcription of type I ISGs (Fig. 1). Upregulated genes include multiple proteins that are themselves RNA sensors and that, as part of an antiviral response, affect RNA homeostasis.
PKR (protein kinase RNA-activated)
PKR is a serine-threonine kinase, encoded by the EIF2AK2 gene. Although PKR is expressed constitutively, it is significantly upregulated by type I and type III interferons (Meurs et al. 1990; Ank et al. 2006). PKR contains two N-terminal double-stranded RNA-binding domains and a C-terminal kinase domain. Two PKR monomers bind dsRNA of at least 30 bp, leading to autophosphorylation and activation of PKR (Lemaire et al. 2008). Viruses use the host translational machinery to replicate, and active PKR phosphorylates eukaryotic initiation factor 2α (eIF2α), which inhibits translation initiation (Meurs et al. 1992). However, the role of PKR in immune responses is manifold and integrated with other RNA-sensing components. In addition to acting as an inhibitor of translation, it is important for IFN-α/β mRNA stability during infection by some viruses (Schulz et al. 2010). PKR also interacts with MDA5 and is important for IRF3 nuclear translocation upon MDA5 activation (Pham et al. 2016).
IFITs (interferon-induced proteins with tetratricopeptide repeats)
The IFITs are cytoplasmic antiviral proteins that, although constitutively expressed, are strongly induced upon viral infection and exert a variety of antiviral effects. The IFITs have different binding specificities for 5′ end motifs. IFIT1 was shown to recognize non-2′-O-methylated capped RNA and inhibit viral translation (Daffis et al. 2010; Habjan et al. 2013). IFIT1 prefers capped but unmethylated RNAs, whereas IFIT5 prefers targets with uncapped 5′-triphosphate ends (Abbas et al. 2013, 2017). While IFIT1 and IFIT2 were initially described as binding eIF3E to inhibit translation initiation (Guo et al. 2000; Hui et al. 2003; Terenzi et al. 2006), newer data suggest that IFIT1 competes with other 5′ cap recognition proteins such as eIF4E and eIF4F, instead of directly associating with the translation machinery, and that interactions with IFIT2 and IFIT3 are important for this function (Pichlmair et al. 2011; Habjan et al. 2013; Kumar et al. 2014; Fleith et al. 2018). Profiling of IFIT5-associated cellular RNAs revealed that tRNAs are one of its major cellular substrates, notably including precursor species. Additionally, many IFIT5-bound RNAs were found to have 3′ oligo(U) tails (Katibah et al. 2014), characteristic of RNAs that are substrates for DIS3L2, a 3′ to 5′ exoribonuclease that functions in the cytoplasmic surveillance of structured noncoding RNAs (Ustianenko et al. 2016).
OAS, RNase L, and OASL
Members of the oligoadenylate synthetase (OAS) family are upregulated by interferons and activated by dsRNA binding to synthesize 2′-5′ oligoadenylates from ATP (Hovanessian et al. 1977, 1979; Kerr et al. 1977; Baglioni et al. 1978; Clemens and Williams 1978). These 2′-5′ oligoadenylates bind with high specificity to RNase L, an endonuclease that is a major effector of the antiviral response. Although monomeric RNase L is inactive, binding to 2′-5′ oligoadenylates results in dimerization (Dong and Silverman 1995; Han et al. 2014). Dimeric RNase L is a potent endoribonuclease that cleaves both viral and host RNAs (Hovanessian et al. 1979; Malathi et al. 2007), leading to a global reduction in protein synthesis, and promoting autophagy and/or apoptosis. The endonucleolytic cleavage products can also be recognized by RLRs, further amplifying the pro-inflammatory response (Malathi et al. 2007).
OASL is related to OAS but lacks 2′-5′ oligoadenylate synthetase activity. It also contains two ubiquitin-like domains in its C terminus. OASL is induced by interferon, binds double-stranded RNA, and enhances RIG-I signaling through its ubiquitin-like domain, possibly because binding of this domain to the RIG-I CARDs mimics their ubiquitination (Zhu et al. 2014).
ADAR1 (adenosine deaminase RNA specific 1)
ADAR1 is an RNA editing protein that binds to double-stranded RNA and converts adenosine to inosine (Bass 2002; Nishikura 2016). Many cellular RNAs can form stable dsRNA structures, including introns and untranslated regions that contain tandem inverted repeats, microRNA precursors, and other noncoding RNAs. The main immunological role of ADAR1 is to reduce activation of innate immune sensors by these endogenous, unedited double-stranded RNAs. In the absence of editing, these RNAs have the potential to be recognized by RNA sensors, and ADAR1 depletion has been shown to increase the activation of MDA5, PKR, and OAS (for review, see Quin et al. 2021). The A- to I-edited RNA can destabilize the dsRNA structure and protect against recognition of self RNA by the sensors described above. There is evidence that ADAR1 negatively regulates PKR independent of its RNA editing function, likely by competitively binding dsRNA (Hu et al. 2023).
EVASION AND ACTIVATION OF RNA SENSORS BY HOST RNAs
In their native and mature forms, most host RNAs are not good substrates for RLRs. MDA5 binds long double-stranded RNA, and RIG-I binds shorter double-stranded sequences (at least 10 bp) with a 5′-triphosphate moiety (Linehan et al. 2018; Ren et al. 2019). For mature, functional endogenous RNAs, many of these features are either unusual or concealed, as the RNAs exist in ribonucleoprotein complexes (RNPs), in which there is little naked RNA. RNA polymerase II transcripts undergo capping at their 5′ end, limiting their ability to be sensed by RIG-I. Although all RNAs synthesized by RNA polymerase III have a 5′-triphosphate moiety, in the case of tRNAs, this is removed upon cleavage of the 5′ leader by RNase P. However, RNAs that fail to mature properly, such as pre-tRNAs that are exported to the cytoplasm for degradation by DIS3L2 (Eckwahl et al. 2015; Ustianenko et al. 2016), or that fail to assemble with protein partners may serve as ligands for some of these RNA sensors. Indeed, during some viral infections, ncRNA processing or localization is altered to generate RIG-I ligands (Chiang et al. 2018; Zhao et al. 2018). For example, during lytic reactivation of Kaposi's sarcoma herpesvirus, transcription of the cellular DUSP11 triphosphatase is reduced, resulting in RIG-I activation through recognition of increased levels of cellular noncoding RNAs containing 5′ triphosphates (Zhao et al. 2018). RLR activation by endogenous ligands has been shown to be limited by the activity of the RNA exosome, a multinuclease complex that is important in the degradation and processing of a broad range of RNA substrates, as depletion of its adapter helicase SKIV2L results in production of type I interferons (Eckard et al. 2014).
Most noncoding RNAs are either nuclear or cytosolic, but the ligand-binding domains of TLRs are within endosomes. Therefore, intracellular RNAs do not generally activate these sensors. However, ribonucleoproteins are common autoantigens in many autoimmune diseases, and immune complexes of antibody and antigen can be endocytosed by immune cells, thereby gaining access to the endolysosomal compartment (Krieg and Vollmer 2007; Marshak-Rothstein and Rifkin 2007). Both the proteases and low pH of endosomal vesicles may destabilize RNA–protein interactions, expose single-stranded RNA, and destabilize RNA secondary structure, promoting TLR engagement.
RNA SENSORS IN SYSTEMIC AUTOIMMUNE AND AUTOINFLAMMATORY DISEASES
Systemic autoimmune and autoinflammatory diseases are diseases in which the immune system is pathologically activated against the host in multiple organs/organ systems. In an autoimmune disease, the adaptive immune system is aberrantly activated, resulting in the production of autoreactive B and T cells, autoantibodies, and concomitant tissue damage, while in autoinflammatory diseases, the innate immune system is activated inappropriately (Szekanecz et al. 2021). Most autoimmune diseases are polygenic, and while individually these diseases are not common, together they are found in as much as 10% of the adult population. The prevalence of autoimmunity (defined by the presence of autoantibodies) and of autoimmune disease has increased significantly in recent decades (Dinse et al. 2022; Conrad et al. 2023; Miller 2023). Over a thousand human autoantigens have been identified, and nucleic acid–binding proteins are the most abundant class of autoantigens (Wang et al. 2017), despite their intracellular (and often nuclear) localization.
Autoinflammatory diseases are a group of disorders in which there is aberrant activation of the innate immune system, leading to inflammation without any preceding trigger. The concept of autoinflammatory diseases emerged from the discovery that mutations to genes in critical pathways of innate immunity could lead to periodic fever syndromes (McDermott et al. 1999). The canonical autoinflammatory diseases are monogenic and rare (Manthiram et al. 2017), and in these diseases, inflammation occurs in an antigen-independent manner (Nigrovic et al. 2020). However, there is significant cross talk between the innate and adaptive immune systems, and while diseases continue to be classified as “autoimmune” or “autoinflammatory,” it is increasingly clear that most systemic diseases of inflammation exist on a continuum, with varying degrees of dysregulation of both the adaptive and innate immune systems (McGonagle and McDermott 2006; Szekanecz et al. 2021). In recent years, mutations in genes that play important roles in RNA sensing and RNA homeostasis have been implicated in both autoimmune and autoinflammatory diseases (Table 1).
Autoimmune and autoinflammatory diseases associated with mutations in RNA sensors and genes important for RNA homeostasis
RNA sensing in systemic lupus erythematosus, an autoimmune disease
Systemic lupus erythematosus is a heterogeneous autoimmune disease (Ghodke-Puranik et al. 2024). The hallmark of SLE is the presence of antinuclear antibodies (Pisetsky and Lipsky 2020). This can include antibodies to dsRNA, as well as snRNP proteins, ribosomal components, and RNA-binding proteins such as Ro60 and La. Clinical presentations are highly variable and can change over time. These manifestations include fever and fatigue, photosensitive skin rashes, arthritis, oral ulcers, cytopenias (low blood cell counts), glomerular (kidney) disease, serositis, and inflammation of the central nervous system (CNS) (Kaul et al. 2016). Autoantibodies precede clinical disease onset by many years (Arbuckle et al. 2003). The earliest detected antibodies are against the Ro60 protein and U1A, a component of the U1 snRNP (Heinlen et al. 2010). These proteins are highly conserved evolutionarily and have bacterial orthologs with high sequence similarity to their mammalian counterparts. While it is not fully understood how tolerance is broken to these antigens, there is growing evidence that autoimmunity to some RNPs, such as the Ro60 RNP, may develop as a consequence of molecular mimicry to their microbial orthologs (Greiling et al. 2018; Williams and Wolin 2021).
The pathogenesis of SLE is complex and involves defective clearance of apoptotic cells, activation of B cells, T cells, and neutrophils, autoantibody production, complement deposition, and type I interferon activation (Crow 2023; Ghodke-Puranik et al. 2024). Aberrant RNA sensing has been implicated in both human studies and animal models of SLE. Genome-wide association studies (GWAS) have found that common polymorphisms in IFIH1 (the gene that encodes MDA5), TLR7, TLR8, and OAS1 are all linked with SLE (Hom et al. 2008; Fike et al. 2019; Yin et al. 2021). More recently, rare gain-of-function variants in several of these genes have been implicated in SLE (Van Eyck et al. 2015; Brown et al. 2022; Natsumoto et al. 2023) (see below).
Mouse models have provided significant insights regarding the role of TLR7 in the pathogenesis of SLE. In a lupus-prone mouse model, deletion of Tlr7 prevented the development of antibodies against RNP antigens and ameliorated lymphocyte activation and renal pathology (Christensen et al. 2006). Duplication of Tlr7 in wild-type mice was sufficient to induce autoantibodies against RNA-containing autoantigens, and a strong gene dosage correlation between Tlr7 and lupus-like autoimmunity was observed. At high levels of Tlr7 expression (increases of 8- to 16-fold compared to wild-type littermates), the mice exhibited uncontrolled regulation of antigen-presenting cells, resulting in a fatal acute inflammatory response (Deane et al. 2007).
Recently, rare gain-of-function variants in TLR7 have been implicated directly in human disease pathogenesis. A Y264H variant in TLR7 was found in a child with severe lupus. The side chain of Y264 forms a hydrogen bond with 2′,3′-cGMP, and modeling of the Y264H substitution predicted a higher binding affinity for cGMP with the histidine substitution. In reporter assays for TLR7 signaling, overexpression of TLR7Y264H led to enhanced signaling compared to overexpression of WT TLR7. Moreover, introduction of the TLR7Y264H allele into wild-type mice was sufficient to induce lupus. TLR7 variants in two other families with SLE were also reported in this study (Brown et al. 2022). Notably, rare gain-of-function variants in UNC93B1, which is required for endosomal trafficking of nucleic acid–sensing TLRs, are also associated with SLE (Al-Azab et al. 2024; David et al. 2024; Rael et al. 2024; Wolf et al. 2024).
Polymorphisms in IFIH1, which encodes MDA5, have also been linked with SLE, as well as other autoimmune disorders such as type I diabetes mellitus, autoimmune thyroid disease, psoriasis, inflammatory bowel disease, ankylosing spondylitis, rheumatoid arthritis, and multiple sclerosis (Martinez et al. 2008; Barrett et al. 2009; Enevold et al. 2009; Ellinghaus et al. 2016; Saevarsdottir et al. 2020). The fact that polymorphisms in IFIH1 are associated with a wide array of autoimmune diseases suggests a broader role in autoimmunity. One SNP that has been identified in many of these GWAS studies results in a nonsynonymous coding variant that is predicted to change an alanine at position 946 to threonine in the IFIH1 C-terminal domain. Human peripheral blood mononuclear cells expressing the IFIH1T946 variant were found to have higher basal and ligand-triggered type I interferon production. Moreover, IFIH1T946 knock-in mice had lower embryonic survival, higher basal IFN-I expression and increased autoimmunity (Gorman et al. 2017). However, because this variant is both common and found in disparate autoimmune diseases, it is unlikely to be sufficient to cause these disorders.
One mutation in MDA5 that has been found to cause both Aicardi-Goutières syndrome (described below) and SLE results in an R779H missense mutation in the helicase domain (Rice et al. 2014; Van Eyck et al. 2015). This residue is close to the ATP-binding site as well as to the protein–protein interface for MDA5 filament assembly. In cell-based reporter assays, this mutation caused increased interferon signaling, even in the absence of ligand stimulation (Rice et al. 2014).
RNA sensing in Aicardi-Goutières syndrome, an autoinflammatory disease
Aicardi-Goutières syndrome (AGS) is a type I interferonopathy (a disease characterized by abnormal activation of type I interferons) and was the first such disease to be described. It is a rare disease: about 500 cases have been reported, and it is caused by mutations in one of several DNA- and RNA-sensing or degradation pathways, including mutations that lead to loss of function of TREX1, a cytosolic exonuclease (Mazur and Perrino 1999); SAMHD1, a dNTP triphosphohydrolase (Chen et al. 2019); ADAR1; the ribonuclease H2 complex, which removes rNTPs that have been misincorporated into DNA (Cerritelli and Crouch 2016) or gain-of-function mutations in IFIH1 (Crow et al. 2015). In a study of 374 patients, over 50% had mutations in a component of the RNase H2 complex, 23% in TREX1, 13% in SAMHD1, 7% in ADAR1, and 3% in IFIH1, although mutations in ADAR1 and IFIH1 may have been underrepresented in this study since the association between these genes and AGS was discovered more recently (Crow et al. 2015). AGS is characterized by high systemic and CNS levels of interferon (Crow and Manel 2015). Onset can be congenital, and the most common feature is CNS inflammation, typically resulting in neurological abnormalities within the first year of life (Crow et al. 2015). The cerebrospinal fluid contains increased levels of lymphocytes and elevated levels of IFN-α. Imaging findings include leukoencephalopathy (inflammation of the white matter) and basal ganglia calcifications. Signs of systemic inflammation, such as fever, can also be present (Crow et al. 2015).
While many features of AGS can be directly attributed to the aberrant activation of type I interferons, AGS is also characterized by extensive autoimmunity. A study of autoantibodies in 56 AGS patients showed that these patients produced a broad spectrum of autoantibodies, including antinuclear autoantibodies and autoantibodies that can target astrocytes and endothelial cells (Cuadrado et al. 2015), which are likely implicated in CNS inflammation. Levels of some antinuclear antibodies (particularly the anti-Sm and anti-RNP antibodies that target spliceosomal snRNPs) were very elevated and higher than in samples from patients with SLE and mixed connective tissue disease. Brain tissue from patients with AGS demonstrated both perivascular immune complex deposition and astrocyte immunoreactivity, suggesting both autoinflammatory and autoimmune contributions to the CNS manifestations in AGS.
Non-CNS clinical autoimmune manifestations of AGS are relatively rare but can affect many different organ systems. Patients with mutations in IFIH1 and ADAR1 are more likely to also develop SLE (Crow et al. 2015). The degree of autoimmunity seen in AGS and other type I interferonopathies, which is not seen in many other autoinflammatory disorders, further emphasizes that aberrant activation of nucleic acid sensors—whether due to gain-of-function mutations causing hyperactive nucleic acid sensor activity or due to the accumulation of aberrant host nucleic acids—is important in the cross talk between innate and adaptive immune regulation.
RNA HOMEOSTASIS AND SENSING IN AGS AND RELATED SYNDROMES
While many of the genetic abnormalities associated with SLE and AGS are in nucleic acid–sensing pathways, mutations in ADAR1 and polynucleotide phosphorylase (PNPase) act by increasing the levels of cellular RNAs that can activate these sensors.
In the initial report of ADAR1-associated AGS, 12 individuals were found to have biallelic variants in ADAR1 that reduced activity (Rice et al. 2012). Seven of the eight amino acid substitutions were in the catalytic domain of ADAR1, many along the dsRNA-binding surface. Various mouse models with homozygous mutations in the catalytic domain of ADAR1 have been established. Although there is significant variability in the severity of the phenotype (depending on whether the mutations retain some editing activity), as with AGS, the mice exhibited elevated levels of ISGs (Liddicoat et al. 2015; Pestal et al. 2015; Inoue et al. 2021). While the most severe mutations result in embryonic lethality, both the lethality and the activation of ISGs were rescued when the mice also lacked MDA5 (Liddicoat et al. 2015; Pestal et al. 2015; Inoue et al. 2021). In a mouse model containing a homozygous mutation that is analogous to a human AGS-associated variant, the mice were viable and fertile but recapitulated both the interferonopathy and CNS inflammation characteristic of human AGS and demonstrated decreased RNA editing in vivo. These phenotypes were abrogated when Ifih1, which encodes MDA5, was also deleted (Inoue et al. 2021). The finding that both gain-of-function mutations in IFIH1 and loss-of-function mutations in ADAR1 result in AGS, coupled with findings in mice that similar phenotypes associated with loss-of-function mutations in Adar1 can be ameliorated by concomitant deletion of Ifih1, supports a model in which mutations that reduce ADAR1 editing result in increased binding of hypoedited host RNAs to MDA5, resulting in aberrant upregulation of interferons and clinical disease.
PNPase-associated interferonopathy
Mutations in PNPase, a 3′ to 5′ phosphorolytic exoribonuclease, can cause an interferonopathy with features of AGS (Vedrenne et al. 2012; Dhir et al. 2018; Bamborschke et al. 2021). PNPase has been well characterized in bacteria, where it functions in multiple degradative processes in the cytosol (Cameron et al. 2018). In mammals, PNPase is found within mitochondria, where its best characterized role is in RNA decay. Because mitochondrial DNA is circular, and RNA synthesis is bidirectional, transcription results in large amounts of double-stranded RNA (Young and Attardi 1975). The role of PNPase and its helicase partner (Suv3) is to degrade the RNA strands that are complementary to mRNAs and functional noncoding RNAs (Borowski et al. 2013; Dhir et al. 2018), although the mechanisms by which the antisense strands are recognized are not well understood. In cells depleted for PNPase, mitochondrial double-stranded RNA levels increase, with some dsRNA released into the cytoplasm (Dhir et al. 2018). This dsRNA is primarily sensed by MDA5, resulting in a type I interferon response (Dhir et al. 2018).
A small number of patients with biallelic loss-of-function mutations in PNPT1, the gene that encodes PNPase, have been described. Although the clinical presentations vary, fibroblasts from at least some of these patients have increased levels of dsRNA in mitochondria and cytosol (Dhir et al. 2018). Consistent with triggering a type I interferon response, these patients had elevated ISGs in their peripheral blood (Dhir et al. 2018; Rius et al. 2019). As the number of patients known to harbor these mutations is currently small, the full clinical spectrum of the disease may not yet be clear.
Notably, a patient meeting the diagnostic criteria for AGS, including elevated ISGs, was recently found by whole exome sequencing to harbor a homozygous mutation in an evolutionarily conserved amino acid near the PNPase active site (Bamborschke et al. 2021). As no other mutations in AGS genes were detected, it seems likely that PNPT1 is yet another gene whose loss of function results in AGS. The identification of PNPT1 as an AGS gene should speed the identification of additional AGS patients carrying these mutations.
There are no FDA-approved treatments for AGS. However, case reports and small observational studies have shown promise for systemic and skin manifestations of AGS with the use of Janus kinase (JAK) inhibitors, which disrupt interferon signaling (Neven et al. 2020; Vanderver et al. 2020; Fremond et al. 2023). The effect of JAK inhibition on CNS manifestations was equivocal, which may be related to the relatively low levels of drug detected in the cerebrospinal fluid. The success of RNA-based therapies for the treatment of several rare genetic neurologic disorders (Anthony 2022) suggests that similar approaches targeting the genetic lesions that cause AGS could lead to more effective therapeutics.
Germline and somatic gain-of-function mutations in TLR8 cause a lymphoproliferative syndrome
While somatic mutations have long been understood to be important in the development of cancers (Ostroverkhova et al. 2023), the role of these mutations in other diseases has not been well characterized, in part because strategies to identify disease-causing mutations frequently filter out low frequency variants. However, in the last five years, several diseases of immune dysregulation have been discovered to be caused by somatic mutations in blood cells (Aluri and Cooper 2023). One such discovery was that gain-of-function mutations in TLR8 cause a disease characterized by elevated pro-inflammatory cytokines, increased susceptibility to infection, bone marrow failure, and lymphoproliferation (INFLTR8; Aluri et al. 2021). The authors reported six unrelated patients with this disease, five of whom had somatic mutations and one of whom had a germline mutation. The somatic mutations were found in myeloid and lymphoid cells, and the mutations affected residues that were important to the conformational change that the TLR8 homodimer undergoes upon activation (Aluri et al. 2021). These mutations resulted in enhanced responsiveness to TLR8 ligands, leading to elevated NF-κB signaling and cytokine production (Aluri et al. 2021).
PERSPECTIVES
Nucleic acid sensing exists at a crossroads between autoinflammation and autoimmunity and is important in mediating communication between innate and adaptive immunity. New diseases at the intersection of autoimmunity and autoinflammation are being discovered. Our knowledge of the genetic and mechanistic underpinnings of more common autoimmune diseases like SLE also continues to expand. The established roles of ADAR1, MDA5, and PNPase in rare interferonopathies point to the importance of correct RNA processing and homeostasis in protecting us from aberrant activation of nucleic acid sensors. In addition to diseases associated with germline mutations, somatic mutations in immune cells have been found to cause diseases of immune dysregulation, including somatic mutations in TLR8 (Aluri and Cooper 2023). However, this may be the tip of the iceberg. Nucleic acid processing and surveillance pathways are manifold, and in coming years, we may discover a much broader role for these pathways in mediating immune functions. This may present novel opportunities for therapeutic interventions.
ACKNOWLEDGMENTS
The authors are supported by the Intramural Research Program of the National Institutes of Health, National Institute for Arthritis and Musculoskeletal Diseases (S.G.W.) and by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (Project ZIA BC011757; S.G.W., S.S., and S.L.W.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080304.124.
-
Freely available online through the RNA Open Access option.
This is a work of the US Government.








