
Trinucleotide repeat expansion and RNA dysregulation in fragile X syndrome: emerging therapeutic approaches
- Program in Molecular Medicine, University of Massachusetts Chan Medical School, Worcester, Massachusetts 01605, USA
- Corresponding author: joel.richter{at}umassmed.edu
Abstract
Fragile X syndrome (FXS) is characterized by intellectual impairment caused by CGG repeat expansion in the FMR1 gene. When repeats exceed 200, they induce DNA methylation of the promoter and the repeat region, resulting in transcriptional silencing of the FMR1 gene and the subsequent loss of FMRP protein. In the past decade or so, research has focused on the role of FMRP as an RNA-binding protein involved in translation inhibition in the brain in FXS model mice, particularly by slowing or stalling ribosome translocation on mRNA. More recent advances have shown that FMRP has a profound role in RNA splicing, at least in some cases by modulating the translation of splicing factor mRNAs. In a surprise, the human FMR1 gene is transcribed in most cases even with a full CGG expansion. However, much of the FMR1 that is produced is misspliced, which can be corrected by splice-switching antisense oligonucleotide (ASO) administration. Other recent findings suggest that inhibition of multiple kinases can demethylate the FMR1 gene and induce the formation of an R-loop in the CGG repeat region, leading to contraction of the repeat and FMRP restoration. These insights are paving the way for possible future therapeutic approaches for this disorder. We highlight the importance of FMRP restoration by ASO-mediated splice switching or CGG repeat modulation as key advances that may lead to successful treatments for FXS.
Keywords
INTRODUCTION
It is now over 40 years since Martin and Bell (1943) discovered the Martin-Bell syndrome, now known as the fragile X syndrome (FXS), as an X-linked heritable disorder characterized by mild to severe intellectual impairment. Ever since, the cause and consequences of FXS have been under intense investigation as evidenced by over 17,000 PubMed Central “hits.” The cause of the syndrome lies in the FMR1 gene, whose discovery in 1991 that it contained a “lengthy CGG repeat” (Verkerk et al. 1991) was transformational for understanding the etiology of this disorder. CGG repeats in the 5′ UTR of FMR1 that are fewer than 55 are not deleterious. However, when the repeats expand to 200 or more, CpG islands that are 5′ to the repeat expansion, as well as the repeat expansion itself, become methylated, which causes transcriptional inactivation and loss of its encoded protein FMRP. FMRP is an RNA-binding protein that binds ∼1000 or more RNAs in the brain, and apparently does so in a cis sequence independent manner. Moreover, FMRP preferentially binds the coding regions of mRNAs (Darnell et al. 2011), which when combined with the observation that it co-sediments with polyribosomes in sucrose gradients (Feng et al. 1997), and that its absence results in (modestly) elevated protein synthesis (Dölen et al. 2007), led to the hypothesis that it slows or stalls elongating ribosomes (Darnell et al. 2011). Many subsequent studies indeed conclude that FMRP, directly or indirectly, stalls or slows ribosome translocation on mRNAs (Darnell et al. 2011; Udagawa et al. 2013; Das Sharma et al. 2019; Shah et al. 2020). At least in the synapto-dendritic compartment of neurons, the inhibitory activity of FMRP is alleviated by its ubiquitination and destruction (Huang et al. 2015), thereby allowing newly synthesized proteins to modify synaptic efficacy, neural connectivity, and higher cognitive function (Huber et al. 2000; Bhattacharya et al. 2012; Richter and Zhao 2021). One confounding issue regarding FMRP is how it slows or stalls ribosomes. A single paper of a structure of reconstituted mammalian ribosomes with partial Drosophila FMRP suggested that it would hinder tRNA and translation factor binding (Chen et al. 2014). However, this study remains unconfirmed a decade on and thus we posit that the mechanism of ribosome stalling by FMRP remains enigmatic.
These and many other observations comprise the now standard molecular model for FXS, which are based primarily on FMRP knockout (KO) mice. Considering that mice do not have a CGG expansion, do not methylate Fmr1, and have varying phenotypes that depend on genetic background (Pietropaolo et al. 2011; Spencer et al. 2011), one can reasonably ask whether we are missing key underlying facets of the disorder and their molecular foundations. The fact that therapeutic approaches based on FMRP KO mice have been disappointing in human clinical trials (Berry-Kravis et al. 2018) may underscore this possibility. Certainly, mouse data have been crucial to understanding FMRP activity, but in this review, we evaluate new evidence based mostly on human FXS tissues and cells and what they could mean for therapies derived from RNA expression/function.
The basics of human FMR1
The 5′ UTR of FMR1 typically harbors ∼30 CGG repeats with a range of 6–55 (Willemsen et al. 2011). Although the mechanism of CGG expansion is still not fully understood, it may involve DNA and RNA secondary structures that form during replication, particularly G-quadruplexes on the leading strand and hairpins on the lagging strand. The G-quadruplex is thought to hinder replication fork progression, causing “slippage” of the replication machinery that facilitates CGG reiteration (Chen et al. 1995). Replication fork stalling can also cause DNA damage and improper DNA repair, which may induce CGG expansion. R-loops formed between nascent FMR1 RNA and its DNA complement leave the single-stranded DNA free to form a secondary structure that promotes DNA damage and instability. These and/or several other molecular events occur primarily during meiosis in female germ cells but perhaps during mitosis and in postmitotic cells as well, all of which may lead to CGG expansion (Zhao and Usdin 2016; Malik et al. 2021; Nobile et al. 2021; Tabolacci et al. 2022).
Two hundred or more CGG repeats in FMR1 comprise a “full mutation,” where the gene is methylated and inactivated. Methylation occurs on CpG islands upstream of FMR1, in the CGG expansion as well as in the intronic region, which in turn drives transcriptional repression through increased tri-methylation on histone H3 lysine 9 (H3K9me3) and reduced methylation on histone H3 lysine 4 (H3K4) (Tabolacci et al. 2005). Transcriptional inactivation of FMR1, however, does not occur on intermediate CGG expansions between 55 and 200. In these cases of the “premutation,” FMR1 RNA is elevated, sometimes quite dramatically (∼10 fold), above that detected in non-FXS cells but a dearth of FMRP (Tassone et al. 2007). The mechanisms underlying the paradoxical increase in FMR1 mRNA, which is caused by elevated transcription and not stabilization of the mRNA, have been studied in relation to the acetylation status of nonmethylated CGG repeats. In premutation carriers, CGG repeat expansion leads to increased histone acetylation, including modifications at H3K9 and H4 near the FMR1 locus, which in turn enhances FMR1 mRNA transcription (Todd et al. 2010). The premutation can result in late-onset disorders such as fragile X tremor ataxia (FXTAS), which is characterized by motor and cognitive impairments (Salcedo-Arellano and Hagerman 2022). The reduction of FMRP may be due to well-conserved upstream near-cognate ACG and GUG translation start sites that are followed by the expanded CGG (Zhang et al. 2022). The CGG expansion G-quadruplex is thought to bind ribosomes via a little understood repeat associated non-AUG (RAN) translation mechanism, leading to polyglycine peptides (FMRpolyG) that form inclusion bodies (Todd et al. 2013), which may be toxic and promote the late-onset neuro-degenerative disorder FXTAS (Sellier et al. 2017).
Splicing dysregulation is a hallmark of fragile X syndrome
As happens so frequently in science, an unexpected result can lead to a major breakthrough. In this case, Shah et al. (2020) were testing the long-held idea that FMRP stalls ribosomes on specific mRNAs, particularly those to which FMRP is CLIPed (crosslink and immunoprecipitation) (Darnell et al. 2011). CLIP is a technique whereby UV light is used to covalently crosslink protein to mRNA, which is followed by immunoprecipitation of the mRNA/protein complex in the presence of denaturing reagents that remove nonspecific adducts. To identify these mRNAs, Shah et al. (2020) treated mouse cortical-hippocampal brain slices with harringtonine, a reagent that “freezes” ribosomes on initiation codons but allows elongating ribosomes to continue transit along mRNA. The slices were then treated with cycloheximide at various intervals to freeze the transiting ribosomes, which was followed by ribosome profiling (Ingolia et al. 2009). Although many mRNAs were bound by stalled or slowly moving ribosomes, they were only mildly coincident with FMRP CLIP targets. Even so, by modifying the ribosome runoff procedure and performing experiments comparing wild type (WT) to FMRP KO mice, Shah et al. (2020) identified ∼50 mRNAs that were associated with FMRP-stalled ribosomes, one of which encoded SETD2, an epigenetic factor that catalyzes H3K36me3. ChIP-seq performed with H3K36me3 antibody showed that this chromatin mark was altered in many genes in the FMRP KO brain; in some cases they were elevated, but in other cases they were reduced. Because H3K36me3 is found primarily in the bodies of genes and has been linked to alternative pre-mRNA processing (Kim et al. 2011), Shah et al. followed up these results by performing RNA-seq and assessing RNA splicing. Amazingly, many RNAs displayed dysregulated splicing, mostly exon skipping. A more thorough follow-up analysis was conducted by Jung et al. (2023), who found that dysregulated splicing occurred not only in various brain regions of the FMRP KO mouse, but in other tissues such as liver and muscle as well. These data suggested not only that the proteome in the FMRP KO mouse is likely to be widely different than normal because of the many mis-splicing events, but also because there was a strong correlation of mis-splicing with the human autistic brain, indicating a molecular convergence between these neurodevelopmental disorders (Shah et al. 2020; Shah and Richter 2021). FMRP may also alter the translation of target RNAs by either interacting with RNA editing enzymes thereby facilitating A to I base changes or via the RNA interference pathways (Caudy et al. 2002; Edbauer et al. 2010; Filippini et al. 2017; Tran et al. 2019).
In a separate study examining FMRP-regulated translation by ribosome profiling with mouse adult neural stem cells, Liu et al. (2018a) uncovered a little known but likely important phenomenon known as translational buffering. That is, in some cases where there were fewer ribosome footprints on an mRNA in FMRP KO cells, the RNA was upregulated to compensate for the reduction in ribosome association, i.e., reduced translation. In a similar vein, the reduction of an mRNA in the KO cells was offset by increased ribosome footprints on that mRNA, again indicating a strong homeostatic response to FMRP deficiency. How this buffering phenomenon occurs is unknown, but points to an incredible cellular plasticity.
RNA mis-splicing is also widespread in human FXS, which was first detected in white blood cells (WBCs) (Shah et al. 2023). However, a shocking result from this analysis was that in 21 of 29 FXS samples, FMR1 RNA was readily detected. Because the cells came from individuals with full CGG expansions who were diagnosed with FXS, these observations seemed to be somewhat contradictory to expansion-driven FMR1 silencing because although several individuals were mosaic, some were not. Even more surprising, most of the FMR1 RNA that was produced was mis-spliced such that exon 1, which contains the CGG expansion and the AUG translation start codon was spliced to a pseudo-exon within intron 1. This mis-spliced RNA, referred to as FMR1-217, is ∼2 kb, is polyadenylated, and could encode a 31 amino acid polypeptide, although whether it does so is unknown. The mis-spliced FMR1-217 RNA accounts for 50%–95% of the total FMR1 transcripts in FXS individuals. Formation of the FMR1-217 RNA, which is also detected in FXS postmortem brain (Shah et al. 2023), is driven by the CGG expansion; FXS cells with 98 triplets do not make FMR1-217, but those with 150 do produce the RNA. How the expansion drives the mis-splice remains under investigation.
Antisense oligonucleotide (ASO) rescue of FMR1 missplicing: a road to a therapy?
Splice-switching ASOs as therapeutic modulators have been spectacularly successful (Crooke et al. 2021), most notably in spinal muscular atrophy (SMA) (Hua et al. 2011) and TDP-43 proteinopathies (Baughn et al. 2023). Following a similar trajectory, Shah et al. (2023) found that at least in FXS lymphoblast cells and fibroblasts, ASOs complementary to regions within FMR1-217 reduced mis-splicing, rescued full-length FMR1 RNA, and restored FMRP to normal levels (Fig. 1). Curiously, ASOs spanning the junction region between FMR1 intron 1 and the pseudo-exon had no effect; only ASOs within FMR1-217 were effective in splice switching. Based on this, it is unclear how the ASOs function to alter splicing.
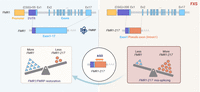
Restoration of FMR1 and FMRP levels by targeting FMR1-217 with ASOs. In typically developing individuals, the FMR1 gene, which contains fewer than 55 CGG repeats and 17 exons, is transcribed and translated to produce the FMRP protein. In FXS, CGG triplets expand to 200 or more, leading to FMR1 gene silencing. In the many cases where the gene is not fully silenced, the repeat expansion results in FMR1 missplicing, producing FMR1-217 in various cells and tissues, including lymphoblastoid cell lines (LCLs), fibroblasts, leukocytes, and brain tissues. The FMR1-217 splicing isoform is composed of exon 1 and a pseudo-exon derived from intron 1. Treatment with ASOs targeting the FMR1-217 pseudo-exon region reduces the accumulation of the FMR1-217 isoform, rescues the production of full-length FMR1 RNA, and restores FMRP protein. Created with BioRender.com.
Nonetheless, the generation of FMR1-217 is clearly dependent on CGG expansion. Liu et al. (2018b) fused inactive Cas9 to TET1 demethylase and used a gRNA to direct the complex to inactive FMR1. TET1 effectively demethylated and activated the gene that contained a full CGG expansion, but analysis of the resulting RNA showed that a prodigious amount was FMR1-217 (Shah et al. 2023). Going further, Vershkov et al. (2019) treated full mutation FXS neural stem cells with 5-azadC, a demethylating agent, which resulted in FMR1 RNA expression, but again, FMR1-217 was produced (Shah et al. 2023). However, treatment of CRISPR/Cas9-edited cells that removed the CGG triplets prior to 5-azadC did not produce FMR1-217. Thus, upstream repeat sequence DNA (i.e., CGG expansion) produces downstream RNA mis-splicing.
In another ASO approach, in this case for FXTAS, Rodriguez et al. (2020) targeted the CGG repeat expansion with the idea that it would prevent translation of the uORFs in this region (i.e., inhibit RAN translation) and preclude the formation of toxic inclusion bodies. Moreover, inhibition of upstream CGG translation might stimulate downstream FMRP synthesis. Indeed, the ASOs successfully reduced (CGG)n translation, suppressed toxicity, and elevated FMRP synthesis. A combination of splice switching and RAN translation-inhibiting ASOs may also be an attractive approach. Thus, ASOs may be a novel therapeutic approach to treat FXTAS and other FXS-related disorders.
Kinase inhibitors, R-loops, and CGG contraction
Early passage FXS human embryonic stem cells (hESCs) produce FMR1 RNA (Colak et al. 2014), an observation that led Lee et al. (2023) to a phenomenal series of experiments that linked protein kinases, R-loops, and CGG contraction at the FMR1 locus. They hypothesized that an extended cell culture that mimics a naive state might induce partial differentiation and epigenetic changes affecting FMR1 expression. Theunissen et al. (2014) had previously shown that culture conditions that included kinase inhibitors targeting MEK, GSK3, BRAF, ROCK, and SRC (so-called 5i) could induce a naive pluripotent state. Lee et al. (2023) found that after 12 days of culture with 5i, FXS-hESCs produced increased FMRP levels that were 50% of those seen in WT iPSCs. This increase was coincident with CGG repeat contraction and demethylation. Notably, no repeat contraction was observed in WT and premutation carrier iPSCs with ∼150 CGG repeats.
Lee et al. (2023) found that kinase inhibition promotes the upregulation of FMR1 demethylation and CGG repeat contraction by inducing endogenous expression of the demethylases TET1 and TET2, which facilitates DNA demethylation through the oxidation of 5-methylcytosine. Site-specific demethylation was achieved using gRNA targeting CGG repeats with dCas9-TET1 reactivated FMR1 and induced CGG repeat contraction. Surprisingly, catalytically dead TET1 also reactivated FMR1 and induced CGG repeat contraction. The authors hypothesize that R-loops that form when the gRNA binds to the FMR1 target site contribute to CGG repeat contraction. Indeed, after culturing cells with 5i, enriched R-loops were detected at the FMR1 transcription start site using DNA–RNA hybrid immunoprecipitation (DRIP). Treatment with a gapmer that disrupts R-loops attenuated CGG contraction, suggesting that R-loop formation is necessary for this process.
How are R-loop formation and CGG repeat contraction related? Does R-loop formation drive CGG contraction, or does CGG repeat contraction and demethylation create space for proteins such as RNA polymerase II, leading to R-loop generation? What is the cause and what is the consequence? To address these issues, Lee et al. (2023) treated FXS iPSCs with dCas9 and CGG gRNA without 5i, revealing that FMR1 reactivation and CGG contraction occurred by inducing R-loops without kinase inhibition, indicating that R-loop formation at the CGG repeats is both necessary and sufficient for FMR1 demethylation and CGG contraction.
How do R-loops cause CGG contraction? Two potential mechanisms for repeat contraction were explored: transcription-coupled nucleotide excision repair (TC-NER) (Lin and Wilson 2007) and the mismatch repair (MMR) pathway (Lin and Wilson 2009). Findings by Lee et al. (2023) indicate that knockdown of the MMR recognition factor MSH2 causes CGG contraction, while knockdown of TC-NER factors does not affect contraction. These results suggest that R-loops recruit DNA repair machinery involved in the MMR pathway, leading to CGG repeat contraction (Fig. 2).
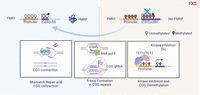
Restoration of FMR1 and FMRP levels through CGG repeat contraction via R-loop formation at the CGG repeat site. Kinase inhibitors upregulate DNA demethylases TET1 and TET2, leading to FMR1 promoter and CGG repeat demethylation. This demethylation initiates FMR1 transcription and facilitates the R-loop formation that is mediated by RNA polymerase II. Subsequent resolution of these R-loops through the MSH2-mediated MMR pathway results in CGG repeat contraction, FMR1 reactivation, and restoration of FMRP levels. Additionally, induction of R-loops using dCas9 and gRNA targeting the CGG repeats also promotes CGG contraction and restores FMR1 and FMRP levels, indicating that R-loop formation at the CGG repeat site is both necessary and sufficient for repeat contraction. Created with BioRender.com.
Therapeutic applications: pros and cons
FXS remains an intractable problem whose treatment is confined to drugs that may mitigate a few symptoms, but not the cause. However, the recent advances noted above in FXS molecular biology that use human, but not animal model systems, suggest a few ways forward. However, it is clear that the road ahead will be a rocky one, which is almost always the case with a new approach to remediate a CNS disorder. Consequently, we point out several obstacles that would need to be considered before moving ahead. For instance, ASO rescue of FMR1 mis-splicing and restoration of FMRP offers a possible new therapeutic advance; even so, major issues remain. For example, how many FXS individuals express FMR1-217 and at what levels in the brain? Would all areas need to take up the ASOs or just, for example, the cortex, hippocampus, and temporal lobes, which are the seats of cognition? As a corollary, how much FMRP is required to rescue FXS pathologies? Then there is the question of how to achieve delivery. As has been done for other CNS disorders (e.g., Tran et al. 2022), intra-thecal injection of ASOs puts them behind the blood-brain barrier where they can travel to the brain and enter at least surface areas of the cortex (Tran et al. 2022). However, some ASOs are not well-tolerated, so if those that are effective in restoring FMRP in cells are somewhat toxic when injected in vivo, this is an obvious problem. Also, there is no animal model for FMR1 mis-splicing, which is a major issue when considering FDA requirements for approval of an experimental drug.
One might envision that the most straightforward therapeutic for FXS would be either CRISPR/Cas9 editing to remove FMR1 CGG triplets or gene demethylation (Park et al. 2015; Vershkov et al. 2019; Hwang et al. 2024). Notwithstanding the difficulty of targeting a gene-editing complex to all or most cells in the brain, or at least the cortex and hippocampus, it has been reported that up to 31 CGG triplets are important for driving FMR1 RNA to dendrites where newly synthesized FMRP protein is thought to be essential (Sirois et al. 2024). Thus, the excision of CGGs that leaves at least 31 triplets but fewer than 55 so as not to cause FXTAS may be a bridge too far for this approach, at least with current technologies. As for DNA demethylation, targeting a protein (demethylase) complex precisely to the FMR1 locus in postmitotic CNS neurons is complicated to say the least. In addition, demethylation induces FMR1 mis-splicing, so this would need to be addressed, perhaps with a splice-switching ASO.
For another therapeutic approach, inducing R-loops in CGG repeats using short gRNAs to lead to repeat contraction might be considered (as in Lee et al. 2023), but CGG repeats in other regions of the genome could lead to off-target DNA excision events. In a similar vein, kinase inhibitors (e.g., 5i) could be problematic due to their likely widespread nonspecific effects on other genes. The mechanisms underlying CGG contraction are not yet fully understood, and precise regulation of the repair system for CGG contraction remains a significant challenge for clinical applications.
Finally, delivery of FMR1 RNA into the CNS via lipid nanoparticles injected into cerebrospinal fluid is plausible (Wong et al. 2012, Tuma et al. 2023, Susa et al. 2024). However, the expression of FMR1 would have to be precise because too much FMRP could lead to deleterious effects by, for example, inhibiting general translation.
In any therapy for FXS, how would one know if it is working? IQ or behavioral analysis would not be an efficient or quantitative readout as one would be concerned with, for example, dosing amount, duration of agent administration, etc. Clearly, what is required is quantitative, statistically robust analysis of biomarkers for the brain. Such biomarkers, whatever they may be, would possibly come from the cerebral spinal fluid (CSF) that bathes the brain. FMRP levels of course would be the best measure of success at the molecular level, but whether it can be detected in CSF at sufficient levels is unknown. Another measure would be FMR1 RNA, but again, it is not clear if it is present in CSF and at what amounts. Biomarkers for FXS therapeutics remain an unmet need.
The challenges of restoring FMR1 expression in the brain are daunting, but in just a few years, enormous strides have been made. Because treating the myriad of downstream symptoms of the disorder has met with very limited success, it seems axiomatic that intervening as high in the FXS molecular hierarchy (i.e., FMR1 gene, RNA, or encoded protein) as possible holds the greatest promise. The coming few years should see refinements in the approaches noted above as well as new ones such as small membrane permeant molecules that alter splicing decisions. Indeed, a large proportion of SMA patients receive Risdiplam, a splice-switching small molecule that is an effective replacement for ASOs (Paik 2022). FXS treatments may follow a similar trajectory.
ACKNOWLEDGMENTS
Work in the authors’ laboratory was funded by National Institutes of Health (NIH) R35 GM149216 and R01 NS132935. We thank Drs. Lori Lorenz and Sneha Shah for reviewing the manuscript.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080270.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








