
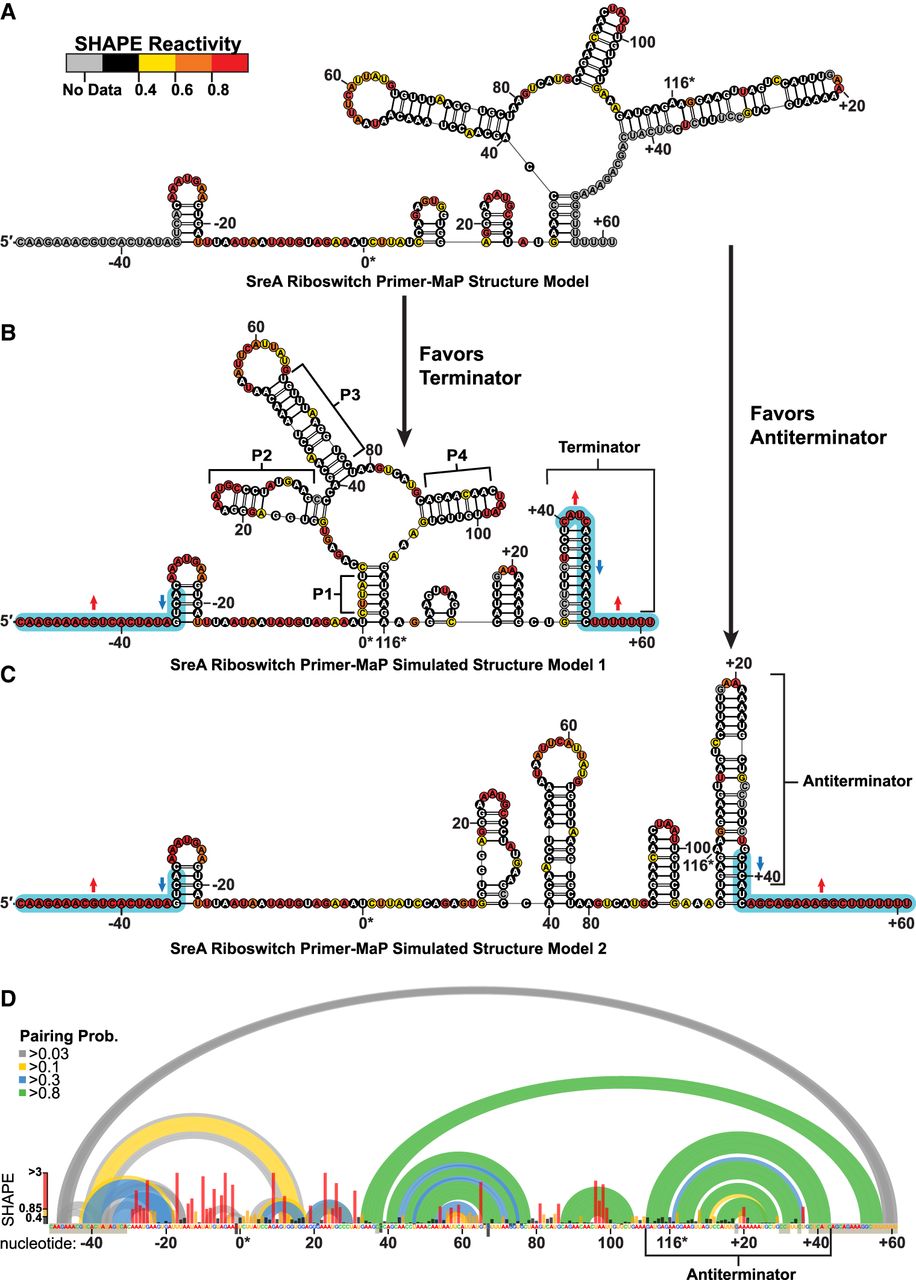
Primer-MaP informed secondary structure model of the full-length SreA riboswitch. (A) The secondary structure model generated from primer-based MaP revealed a structure incongruent with the expected SAM-I fold due to the loss of reactivity in the 3′ end from primer masking. (B,C) Simulated reactivity data, indicated by the blue highlights, generate expected terminator (B) or antiterminator (C) conformations by modeling. (D) The pairing probabilities (colored arcs) of the SreA riboswitch derived from SHAPE-informed partition function. Normalized SHAPE reactivities are plotted (bar graph) per nucleotide, along with primer masked or high background sites (gray boxes over nucleotides). The antiterminator helix is the predominant fold in the structural ensemble, nt 110 to +43. Numbering is oriented to the native aptamer sequence (nt 0–116).








