
Development of bioconjugate-based delivery systems for nucleic acids
- Aniket Wahane1,4,
- Vishal Kasina1,4,
- Mounika Pathuri1,
- Ciara Marro-Wilson2,
- Anisha Gupta2,
- Frank J. Slack3 and
- Raman Bahal1
- 1Department of Pharmaceutical Sciences, University of Connecticut, Storrs, Connecticut 06269, USA
- 2Department of Pharmaceutical Sciences, University of Saint Joseph, West Hartford, Connecticut 06033, USA
- 3Department of Pathology, HMS Initiative for RNA Medicine, BIDMC Cancer Center, Harvard Medical School, Boston, Massachusetts 02115, USA
- Corresponding author: raman.bahal{at}uconn.edu
-
↵4 These authors contributed equally to this work.
Abstract
Nucleic acids are a class of drugs that can modulate gene and protein expression by various mechanisms, namely, RNAi, mRNA degradation by RNase H cleavage, splice modulation, and steric blocking of protein binding or mRNA translation, thus exhibiting immense potential to treat various genetic and rare diseases. Unlike protein-targeted therapeutics, the clinical use of nucleic acids relies on Watson–Crick sequence recognition to regulate aberrant gene expression and impede protein translation. Though promising, targeted delivery remains a bottleneck for the clinical adoption of nucleic acid-based therapeutics. To overcome the delivery challenges associated with nucleic acids, various chemical modifications and bioconjugation-based delivery strategies have been explored. Currently, liver targeting by N-acetyl galactosamine (GalNAc) conjugation has been at the forefront for the treatment of rare and various metabolic diseases, which has led to FDA approval of four nucleic acid drugs. In addition, various other bioconjugation strategies have been explored to facilitate active organ and cell-enriched targeting. This review briefly covers the different classes of nucleic acids, their mechanisms of action, and their challenges. We also elaborate on recent advances in bioconjugation strategies in developing a diverse set of ligands for targeted delivery of nucleic acid drugs.
Keywords
INTRODUCTION
In recent years, nucleic acid therapeutics have seen a resurgence with several FDA-approved drug products and promising candidates in clinical trials. The popularity of nucleic acid therapeutics stems from the versatility of RNA-based drugs to modulate target gene and protein expression, thus demonstrating enormous potential to treat various genetic and rare diseases (Roberts et al. 2020; Kulkarni et al. 2021). The accomplishment of the Human Genome Project (Lander et al. 2001; Nurk et al. 2022) has allowed the identification of target sequences of genes of interest, resulting in a streamlined drug development process of RNA-based drugs (Gibbs 2020). Nucleic acid-based therapeutics consist of antisense oligonucleotides (ASOs) and small-interfering RNAs (siRNAs) that inhibit aberrant protein translation by targeting messenger RNA (mRNA). ASOs are single-stranded oligonucleotides that bind to RNA target sequences by Watson–Crick base-pairing and inhibit their gene expression by recruiting the RNase H enzyme (Crooke et al. 2018). siRNAs are double-stranded oligonucleotides that induce RNA interference (RNAi) when the guide strand forms a complex with Argonaute 2 (Ago2) enzyme, which then screens target mRNA sequence by Watson–Crick recognition and in the presence of a 100% complementary mRNA target, results in mRNA cleavage.
Though proteins have been traditionally targeted for drug development, the lack of binding pockets on many target proteins makes them undruggable (Shen and Dassama 2023). Thus, RNA-based nucleic acid therapeutics are an attractive strategy for developing novel therapeutics for undruggable targets.
This review provides a brief overview of therapeutic oligonucleotides, their mechanism of action, progress made in recent years, and the challenges associated with their targeted delivery for broader clinical translation. We further discuss inclusively the advances in covalent bioconjugation strategies for delivering nucleic acids modalities for RNA therapeutics.
ANTISENSE MECHANISM OF ACTION OF NUCLEIC ACIDS
Based on their mechanism of action, ASOs can be classified into two categories: (i) cleavage-based and (ii) non-cleavage-based oligonucleotides. Phosphorothioate-modified single-stranded ASOs (PS-ASOs) and siRNA belong to mRNA cleavage-based oligonucleotides as they induce mRNA cleavage by RNase H1 or RNAi, respectively. Overall, a key difference between nucleic acids (PS-ASO and siRNAs) can be attributed to the mechanism of action of their associated nucleases, RNase H and Ago2. The mechanism of action for the cleavage of mRNA for ASOs follows the binding of the ASO sequence to the target mRNA before RNase H recognition and cleavage. On the other hand, for siRNAs, the Ago2 enzyme first forms a complex with the guide or antisense siRNA strand and later interacts with the target mRNA sequence.
On the other hand, phosphorodiamidate morpholino oligomers (PMOs), peptide nucleic acids (PNA), and locked nucleic acids (LNA) are non-cleavage-based nucleic acids. PMOs are splice-switching ASOs that bind to the pre-mRNA and either inhibit or promote exon inclusion to modulate gene expression. Similarly, PMOs, PNA, and LNAs can act as steric structural blockers and inhibit the interactions of specific mRNAs with ribosomal assembly to cause translational arrest. Another key difference between different ASO classes is their site of action in the cell. Splice-switching ASOs operate in the nucleus, whereas PS-ASOs, siRNAs, LNAs, and PNAs can act in the nucleus or cytoplasm. This review provides a brief overview of the chemistry and mechanism associated with the aforementioned nucleic acids.
RNAi-inducing oligonucleotides
siRNAs are short (20–24 nt) double-stranded sequences that engage the native endogenous cellular defense RNAi mechanism to target specific mRNA. The discovery of the RNAi mechanism (Fire et al. 1998) opened avenues for siRNAs to target transcription factors and dysregulated genes associated with various disease pathologies. siRNAs induce RNAi by Watson–Crick recognition of the target mRNA sequences by assembling a siRNA guide strand RNA-induced silencing complex (RISC) comprising Ago2 proteins. The siRNA guide strand then scans for target mRNA sequences and directs the RISC complex to a perfectly complementary mRNA sequence wherein Ago2 protein cleaves the target mRNA. As of 2024, the US FDA has approved six siRNA drugs: Patisiran (Onpattro) (Adams et al. 2018), Givosiran (Givlaari) (Scott 2020), Eplontersen (Oxlumo) (Scott and Keam 2021), Inclisiran (Leqvio) (Lamb 2021), Vutrisiran (Amvuttra) (Keam 2022), and Nedosiran (Rivfloza) (Syed 2024).
RNase H-activating oligonucleotides
PS-ASOs recruit RNase H1 to initiate the target mRNA cleavage. PS-ASOs contain phosphate groups wherein the nonbridging oxygen atom is replaced by a sulfur atom, increasing their enzymatic resistance (Stein et al. 1988) and plasma binding (Gaus et al. 2019; Crooke et al. 2020). PS-ASOs have optimal stability in plasma and, following systemic circulation, exhibit moderate liver retention due to internalization by stabilin-1 and stabilin-2 (STAB1 and STAB2) scavenger receptors present on liver sinusoidal epithelial cells (Miller et al. 2018a). In particular, gapmer design-based ASOs have gained much attention as an attractive therapeutic modality. Gapmer design consists of a central region of unmodified nucleotides (8–10 mer) flanked by modified nucleotides to facilitate RNase H1-mediated cleavage activity, whereas flanking modified nucleotides enhance its binding affinity and enzymatic resistance properties (Zhang et al. 2021). Currently, there have been six FDA-approved PS-modified ASO drugs: Fomivirsen (Vitravene) (Roehr 1998), Mipomersen (Kynamro) (Wong and Goldberg 2014), Heplisav-B (Lee and Lim 2021), Inotersen (Tegsedi) (Keam 2018) (Benson et al. 2018), Volanesorsen (Waylivra) (Paik and Duggan 2019), and Tofersen (Qalsody) (Blair 2023).
Splice-switching oligonucleotides
PMOs belong to splice-switching ASOs wherein the five-membered backbone sugars are replaced by six-membered morpholine subunits interconnected with phosphorodiamidate linkages (Summerton and Weller 1997). PMOs are single-stranded nucleic acid analogs that offer several advantages, such as high solubility, resistance to enzymatic degradation, and optimal binding affinity due to their neutral morpholino backbone (Maksudov et al. 2023). PMOs designed to target pre-mRNA regions can be widely used for exon-skipping-based applications. The exon-skipping applications for PMOs are exclusively used to target rare diseases such as Duchene muscular dystrophy (DMD), where selective exons can be skipped to restore the open reading frame for translation of functional shortened dystrophin protein (Popplewell et al. 2009). Currently, there are four FDA-approved drugs for the treatment of DMD, namely, Eteplirsen (Exondys 51) (Syed 2016), Golodirsen (Vyondys 53) (Heo 2020), Viltolarsen (Viltepso) (Dhillon 2020), and Casimersen (Amondys) (Shirley 2021). Nusinersen, a 2′-O-methoxyethyl (MOE) PS-ASO, is used for treating spinal muscular atrophy (SMA) caused by deficiency in the survival motor neuron-1 (SMN1) protein due to a mutation in the chromosome 5q. A second SMN gene, SMN2, is not able to compensate because its pre-mRNA is normally misspliced. Nusinersen increases exon 7 inclusion in the SMN2 mRNA transcripts by displacing the intronic splice silencing site 1 (ISS-1) between exons 7 and 8, increasing SMN protein production (Hua et al. 2008; Finkel et al. 2016; Bennett et al. 2019). Similarly, Milasen is a patient-specific 2′-MOE-based PS-ASO that rescues the splicing of neuronal ceroid lipofuscinosis 7 (CLN7) in Batten's disease by a splicing modulation mechanism (Kim et al. 2019).
Steric-blocking oligonucleotides
LNAs contain a methylene bridge group between the 2′ oxygen and 4′ carbon groups of the ribose sugar, thus locking the ribose sugar moiety in a particular conformation. This results in the A-helical conformation after LNAs bind to complementary DNA/RNA target sites (Braasch and Corey 2001; Obika et al. 2001). LNAs offer several advantages, such as higher binding affinity to their complementary targets (Tm increase by 3°C–9°C per LNA modification) (Singh and Wengel 1998) due to their preorganized structure and resistance to enzymatic degradation. In addition, LNAs have extended circulation and tissue half-life after systemic delivery (Kurreck et al. 2002; Frieden et al. 2003). LNA-based ASOs utilize either a gapmer (as explained in the above section) or mixmer design to exert their antisense activity. The mixmer design contains interspaced DNA- and LNA-based nucleotides. Mixmer LNAs induce steric-blocking mechanisms to impede mRNA translation (Braasch and Corey 2001; Hagedorn et al. 2018).
PNAs are synthetic analogs of nucleic acids that contain a neutral 2-amino ethyl glycine backbone, replacing the negatively charged phosphodiester backbone with nucleobases connected via a methyl carbonyl linker (Egholm et al. 1992). The neutral backbone of PNAs offers high binding affinities to their complementary DNA/RNA targets and resistance to enzymatic degradation, making them potential therapeutic agents for antisense applications (Hanvey et al. 1992). PNAs perform their antisense effect as steric blockers as they interfere with mRNA translation by preventing its binding with the ribosomes (Knudsen and Nielsen 1996). At present, one PNA-based drug is being investigated as a pain medication in osteoarthritis patients (NCT05216341). Similarly, various chemically modified gamma PNAs have been developed that can target the RNA effectively by stronger binding affinity than conventional PNAs. Gamma PNAs are preorganized into a right-handed helical conformation, which contributes to their strong binding affinity with the complementary RNA target sites (Dhuri et al. 2021, 2023; Wang et al. 2023; Pradeep et al. 2024). Antisense PNA-based fluorescence in situ hybridization (FISH) probes have also received FDA approval as diagnostic agents to detect bacterial pathogens in blood cultures (Radic et al. 2016; MacLelland et al. 2024).
Emerging chemical modifications
To broaden the therapeutic potential of nucleic acids, several chemical modifications have been investigated to improve their properties, making them a more effective and safer therapeutic modality. Excellent review articles have reported a detailed overview of different nucleic acid-based chemistries and structures of oligonucleotides (Shen and Corey 2018; Egli and Manoharan 2023).
Emerging technologies have also focused on generating stereopure oligonucleotides and exploring other phosphate backbone modifications. The synthesis of PS-modified ASOs generates two stereoisomers (pro-Sp and pro-Rp) with different configurations, which results in racemic mixtures that result in different pharmacokinetic and pharmacodynamic properties (Iwamoto et al. 2017; Kandasamy et al. 2022b; Genna et al. 2023). Thus, stereopure backbones are now being incorporated to mediate the potency and durability of silencing (Liu et al. 2023). Recent reports have highlighted improved duration of action, nuclease stability, and low inflammatory profile of mesyl phosphoramidate (MsPA) modified ASOs compared to standard PS-ASOs (Miroshnichenko et al. 2019; Patutina et al. 2020; Anderson et al. 2021). The introduction of a phosphoryl guanidine (PN) backbone showed increased potency (∼10-fold) and distribution in CNS compared to the PS backbone (Kandasamy et al. 2022a). PN- and MsPA-modified oligonucleotides replace the oxygen atom in the phosphodiester backbone with a phosphoryl guanidine or methanesulfonyl group, respectively, and exert their antisense action by recruiting RNase H to cause target mRNA degradation.
THE DELIVERY CHALLENGE FOR RNA THERAPEUTICS
For the success of RNA therapeutics, efficient organ-selective delivery and cellular uptake remain enormous challenges. Before the nucleic acid cargo undergoes cellular uptake, it must overcome several barriers, such as susceptibility to serum nuclease degradation, immune cell activation, and nonselective targeted delivery. Immune cell activation by Toll-like receptors can be reduced by chemical modifications of nucleic acids. In particular, most approved and later-stage clinical siRNAs are fully chemically modified, which reduces their recognition by toll-like receptors (Karikó et al. 2005; Sioud 2005; Meng and Lu 2017).
To attain therapeutic efficacy, nucleic acids are administered systemically at high doses, often leading to increased side effects. Nucleic acids are primarily retained in the liver and kidney (Geary et al. 2015). It has been established that the unwanted accumulation of ASOs in the kidney can cause nephrotoxicity (Frazier 2022; Wu et al. 2022). In addition, partial sequence homology in other off-target sites can result in nonspecific binding of the ASOs and altered expression of undesired genes (Yoshida et al. 2019).
The systemic toxicity of ASOs and siRNAs mainly depends on their pharmacokinetic properties, which are influenced by several factors, including chemical structure and molecular weight. Unconjugated ASO toxicity increases with the length (or molecular weight) of the ASO sequence (Aupy et al. 2020), whereas for conjugated siRNAs, the dose and administration route also affect the pharmacokinetic-based toxicity (Sten et al. 2023). In CNS applications, Tominersen, an ASO targeting the Huntingtin (HTT) protein, was administered intrathecally in clinical trials (Kingwell 2021). However, the trials had to be halted as hydrocephalus was observed in patients.
Several FDA-approved drugs carry warnings for hepatotoxicity and nephrotoxicity. Drugs such as Mipomersen and Inotersen exhibit black box labeling for hepatotoxicity and nephrotoxicity (Wu et al. 2022), whereas Viltolarsen has shown nephrotoxicity during preclinical studies (Freed 2020).
Further, nucleic acids must generally undergo substantial cellular uptake to show their antisense activity. It has been demonstrated that the cellular uptake of nucleic acids alone is governed predominantly by clathrin-mediated endocytosis (Juliano et al. 2014). However, nucleic acids undergo substantial endosomal entrapment during endocytosis, limiting their availability to the target RNA. Hence, endosomal release of nucleic acids is imperative to reach the target site in the cytoplasm. For example, PS-ASOs are reported to be transported by the Rab5-mediated endosomal route postinternalization by stabilin receptors, and the endosomal escape is facilitated by Rab7 and lysobisphosphatidic acid (LBPA) in late endosomes (Miller et al. 2018b). Ligand-conjugated ASOs such as N-acetyl galactosamine-conjugated ASOs (GalNAc-ASOs) show ∼1%–2% endosomal release in vivo in hepatocytes (He et al. 2021). Since GalNAc-siRNAs conjugates show ∼0.3% cytoplasmic availability in vivo (Brown et al. 2020), this reveals that ∼0.3%–2% of nucleic acid cargoes can escape the endosomes. Similarly, lipid nanoparticle (LNP) delivered siRNA undergoes up to 70% exocytosis through late endosomes. Niemann-Pick type C1 (NPC1) is a major recycling pathway for LNP-delivered siRNA (Sahay et al. 2013). LNP-delivered siRNA shows increased cellular retention and gene silencing activity in NPC1-deficient cell lines.
Therefore, nucleic acid cargoes must undergo optimal cellular uptake followed by endosomal escape to interact with their therapeutic RNA targets for effective therapy (Fig. 1). Various endocytic small molecule agents, cationic peptides, lipid conjugates, and viral proteins (Dowdy 2023) have been used to increase nucleic acids’ endosomal escape. Similarly, various strategies involving cationic carriers (Wahane et al. 2020), receptor-targeted ligands (Srinivasarao and Low 2017), transporter-mediated entry (Zhou et al. 2015), and cell-penetrating peptides (CPPs) (Gori et al. 2023) have been employed to overcome the above-mentioned extracellular and intracellular barriers. We will next cover the bioconjugates for the delivery of nucleic acids.

Challenges for nucleic acid delivery. Schematic depicting major challenges and barriers that impede the clinical translation of nucleic acids.
BIOCONJUGATION STRATEGIES TO OVERCOME THE DELIVERY CHALLENGE
Cell-penetrating peptides
To increase cellular delivery, various ASOs have been covalently conjugated with CPPs, which act as transmembrane peptides to transport ASOs across the cell membrane. In particular, CPPs have a high potential for delivering ASOs into cells and have been conjugated with neutral ASOs like PMOs and PNAs (McClorey and Banerjee 2018).
CPPs derived from the protein transduction domain of the human polyhomeotic 1 homolog transcription factor (known as DG9 peptide) have been used to deliver PMOs in CNS for muscular dystrophy (Wesolowski et al. 2011). DG9-conjugated PMOs had an optimal CNS distribution that rescued early respiratory dysfunction and led to improved survival in a severe SMA mouse model after subcutaneous administration (Aslesh et al. 2023). The delivery potential of DG9 peptide has also been explored for DMD by targeting multiexon skipping (45–55) using PMO combinations, resulting in dystrophin protein restoration and muscle function in hDMDdel52;mdx mice (Lim et al. 2022).
Similarly, PNAs-CPP have been tested for their antiviral (Shehzadi et al. 2023) and antibacterial activity (Goltermann et al. 2019; Tsai et al. 2023). PNAs-CPP conjugates have been employed to effectively target small RNA (sRNA) of the bacterial cell wall to block the mechanisms that confer antibiotic resistance. Using cell-free transcription–translation reactions, a sequence of 10–12 base pairs was identified that binds with the MHC class I polypeptide-related sequence F (MicF) sRNA in controlling the expression of the bacterial outer membrane protein (also known as OmpF). Anti-MicF 12 and anti-MicF 33 PNAs conjugated to amino acid units containing (KFF)3K peptide showed optimal inhibition of MicF activity when combined with nalidixic acid treatment (Tsai et al. 2023). PNAs conjugated to (KFF)3K peptide also resulted in optimal efficacy at micromolar concentrations by targeting chromosome replication initiator protein DNA sequences on the oriC of Escherichia coli bacteria, effectively inhibiting DNA replication (Campion et al. 2024).
siRNA-PF14 conjugates using CPPs called PepFect14 (PF14) comprising stearyl-transportan10 (stearyl-TP10) have been used to cross the blood-brain barrier and suppress glioblastoma cell growth (Srimanee et al. 2018). In another study, CPPs of buforin IIb (BR2) were synthesized and conjugated to siRNA targeting vascular endothelial growth factor (VEGF). BR2 is a 17-amino acid antimicrobial peptide that has demonstrated cancer cell specificity (Cho et al. 2009). A BR2-siRNA results in a 10%–25% increase in transfection efficiency compared to naked siRNA transfection (Lee et al. 2018). The CPPs thus offer various advances in targeting mRNA when they are covalently conjugated to ASOs. However, their clinical translation is still impeded because of insufficient in vivo stability, toxicity, and endosomal escape (Khairkhah et al. 2023).
In particular, the toxicity of CPP-conjugated ASOs can be influenced by the charge of the peptide and the cargo being delivered. TP10 is a CPP that has been known to cause toxicity. TP10, in combination with double-stranded DNA, results in a substantial decrease in its toxicity (El-Andaloussi et al. 2007). Artificial intelligence and machine learning tools can be used to predict the biocompatibility of different CPPs to deliver different classes of ASOs and circumvent their potential toxicity issues. A library of CPP-PMO was constructed and validated using a fluorescence reporter assay (Wolfe et al. 2018). A range of CPPs was used, including arginine-rich peptides with different physicochemical properties. Based on the computational model, it was established that more than three arginine residues can contribute to the in vivo toxicity of CPP-conjugated PMOs. Artificial intelligence and machine learning are valuable tools for high-throughput screening of a broad class of CPP-conjugated ASOs.
In addition to CPPs, chemically engineered peptides such as pH-low insertion peptides (pHLIP) have been used to deliver cargo across the cell membrane into an acidic tumor microenvironment (Wyatt et al. 2018). The translocation of the pHLIP peptide across an acidic membrane is mediated by the protonation of aspartic and glutamic acid residues, which provides hydrophobicity and results in pHLIP folding into an alpha helix, which eventually allows the pHLIP to partition across membranes (Slaybaugh et al. 2020). Tumor-targeted delivery of pHLIP-siRNA conjugates targeting carcinoembryonic antigen-related cell adhesion molecule 6 results in ∼35% tumor reduction in a lung adenocarcinoma xenograft mouse model following systemic administration (Son et al. 2019). pHLIP has been used to target oncogenic microRNA-155 in lymphomas using regular PNAs (Cheng et al. 2015) and gamma PNAs (Dhuri et al. 2023). In addition, different sizes of PNAs (up to 7 kDa) undergo biodistribution and accumulation in the tumors via pHLIP-mediated delivery after systemic delivery in vivo (Svoronos et al. 2020). pHLIP has also been used to deliver PNAs targeted against the undruggable DNA double-strand break repair factor KU80 for radio sensitization (Kaplan et al. 2020).
Similarly, 30 amino acid-containing peptides, known as RALA peptides, have also been well-documented for enhanced delivery of siRNA targeting the FKBP prolyl isomerase-like (FKBPL) mRNA in vivo (Bennett et al. 2015). RALA peptides also undergo uptake in pH-dependent conditions. At low pH (pH 5.0), RALA peptides undergo an α-helix conformational change, leading to cytosolic delivery of the cargo (McCarthy et al. 2014). There have also been advances in the development of RNA targeting using siRNA tumor-penetrating nanocomplexes (TPN). Tumor-penetrating peptides such as LyP-1, conjugated to siRNA, have shown tumor-specific accumulation, resulting in DNA-binding 4 (ID4) gene silencing in the tumors (Ren et al. 2012).
Lipid-based ligands
Lipid-based delivery systems are commonly used to deliver ASOs due to their potential to encapsulate negatively charged nucleic acids. In general, negatively charged nucleic acids can readily form lipoplexes with cationic lipids due to ionic interactions and, thus, have been widely used as transfection agents. Although cationic lipids can exhibit good in vivo performance, their excess positive charge is usually associated with immunogenicity and systemic toxicity. Moreover, identifying critical quality parameters such as pH, temperature, and physical state, which impact the long-term stability of lipid-based nanocarriers, is vital for a smooth clinical translation. On the other hand, lipid-based bioconjugates are more promising as they significantly lower the lipid burden-associated toxicity, provide high-dose delivery, and offer less batch-to-batch variation.
Cholesterol, an essential component in mammalian cells, is a commonly used lipid in lipid-based nanocarriers. It provides a structural framework for the cell membrane and significantly impacts its fluidity and thickness, thereby affecting membrane fusion and endocytosis pathways (Yang et al. 2016). Cholesterol as a targeting moiety has been explored to improve cellular transfection, facilitate tissue penetration, and provide serum stability for siRNA therapeutics (Rossi 2004).
The first use of cholesterol as a structural modification for nucleic acids was explored two decades ago when cholesterol-conjugated ASOs administered intravenously exhibited approximately twofold higher hepatic uptake in rats than the nonconjugated controls (Bijsterbosch et al. 2000). This led to further exploratory studies to identify additional lipidic moieties for efficient liver targeting to develop therapies against hepatic diseases. A lipid-based screen of different siRNA conjugates comprising derivatives of lithocholic acid, lauric acid, and cholesterol led to the identification of cholesterol-based siRNA conjugates, which exhibited up to 40% higher silencing than other lipid derivatives (Lorenz et al. 2004). The siRNA design was further optimized by incorporating a PS backbone, 2′-O-methyl sugar modifications on the sense and antisense strands, and cholesterol on the 3′ end of the sense strand. The therapeutic silencing of the apolipoprotein B (ApoB) gene in a transgenic mouse model following intravenous administration was studied using the aforementioned design template. ApoB silencing by cholesterol-conjugated siRNAs (chol-siRNA) considerably reduced plasma ApoB levels and total cholesterol, thus highlighting the value of cholesterol conjugation in improving in vivo pharmacological efficacy of siRNA (Soutschek et al. 2004).
Further investigations highlighted mechanisms for improved in vivo silencing by chol-siRNA conjugates. Tissue-specific accumulation of chol-siRNA is dependent on interactions with lipoprotein receptors, such as low-density lipoprotein, which directs siRNA cargo to the liver, gut, and kidney. However, high-density lipoprotein directs siRNA predominantly to the liver (Wolfrum et al. 2007). Chol-siRNA has also been explored to target liver macrophages to treat nonalcoholic fatty liver disease (NAFLD). In another study, chol-siRNA targeted tumor necrosis alpha (TNF-α) mRNA. The potency of chol-siRNAs led to the complete ablation of TNF-α mRNA, which reduced steatosis, inflammation, and fibrosis in an NAFLD mouse model (Craig et al. 2023).
Recent advances in cholesterol-mediated delivery of siRNAs are centered on extra-hepatic tissue targeting. In preclinical studies, chol-siRNAs have been explored for dendritic cell silencing in the vaginal tissue by intravaginal delivery (Basar et al. 2023). By silencing different host factors that govern viral uptake and dendritic cell migration, a better understanding of the immunogenic function of DCs and various pathways of vaginal HIV transmission could be explored.
Delivery of ASOs to dystrophic muscle remains a crucial hurdle for developing therapies for rare diseases such as muscular dystrophy, due to the presence of fibrotic tissue. Chol-siRNAs are promising candidates for achieving muscular distribution in vivo. Although delivery of siRNAs to the skeletal muscle had been established earlier (Khan et al. 2016), further studies demonstrated the efficient silencing of activin receptor type-1B (Alk4) mRNA, which is a key component in the myostatin signaling pathway, using chol-siRNAs in the mdx mouse model for DMD (Engelbeen et al. 2023).
A study found that PS-ASOs conjugated with cholesterol were toxic in mice and were not further tested in nonhuman primate models (Østergaard et al. 2019). Recently, preclinical studies have been performed to evaluate the efficacy and toxicity of chol-siRNAs. Histopathological analysis showed no adverse effects in the major organs after the intra-articular injection of chol-siRNAs (Lin et al. 2023). Hence, it will be interesting to investigate further the toxicity associated with the cholesterol-conjugated ASOs and siRNAs.
Recently, the albumin binding potential of siRNA conjugates-based methods has been explored to facilitate tumor targeting. Using a library of siRNA-lipid conjugates, exploratory studies were performed to identify lipids that are associated in situ with albumin, contributing to intracellular delivery of cargoes to the tumors. Intravenous delivery of siRNA conjugated with ethylene glycol-based C18 divalent lipid demonstrated an ∼12-fold increase in accumulation relative to unconjugated siRNA in orthotopic triple-negative breast cancer tumors. Moreover, 80% silencing of the gene for myeloid leukemia cell differentiation protein (MCL-1) was seen, leading to better survival outcomes than MIK665 (MCL-1 small molecule inhibitor) in triple-negative breast cancer mouse models (Hoogenboezem et al. 2024).
FDA-approved siRNA therapies include those that silence the transthyretin (TTR) gene in liver for hereditary transthyretin amyloidosis (ATTR) therapy. Higher amyloid deposition in other organs and tissues, such as the eye, often leads to ocular amyloidosis, which is responsible for vitreous opacity and glaucoma. Hence, lipophilic 2′O-hexadecyl (C16) lipid-based siRNA conjugates containing a short lipid chain have been investigated for ocular delivery. A library of C16 lipid siRNA conjugates was screened, and an ∼50% decrease in humor TTR levels postintravitreal siRNA administration in rabbits was noticed for the lead siRNA conjugate (Watanabe et al. 2024).
Carbohydrate-based ligands
Carbohydrates have gained traction for their application in organ-specific nucleic acid delivery (Fig. 2). They possess several advantages, including simple conjugation steps and a low toxicity profile due to their biodegradability. Carbohydrate-conjugated nucleic acid payloads undergo receptor-mediated endocytosis. It has been well established that the GalNAc ligand targets hepatocytes through the asialglycoprotein receptors (ASGPR). ASGPR was the first lectin-type receptor to be discovered in mammals (Grozovsky et al. 2015). Glycoproteins containing D-galactose or N-acetylgalactosamine (GalNAc) have a high affinity for ASGPR. The therapeutic potential of siRNAs was first explored using a trivalent GalNAc moiety for efficient gene silencing in vivo via hepatocyte-mediated delivery (Matsuda et al. 2015). GalNAc-siRNA conjugates were chemically modified to silence the TTR mRNA levels in vivo. At a 5 mg/kg dose of the conjugated siRNA for 72 h, circulating TTR mRNA levels were significantly reduced compared to the control-treated mice. The safety assessments also indicated that the conjugates had no adverse effects during in vivo rat studies (Janas et al. 2018). Studies have also shown that altering GalNAc chemistry can improve the targeting of ASGPR (Weingärtner et al. 2020). Serial assembly of monovalent GalNAc with a 5-hydroxypentanoic acid tether between the sugar moiety and the linker shows efficient delivery of siRNA to the liver, which can reduce the cost and simplify the conjugation steps (Li et al. 2024). GalNAc-siRNA conjugates have been utilized to reduce the levels of Angiopoietin-like protein 3, which is upregulated in dyslipidemia and cardiovascular disease (Wang et al. 2023).
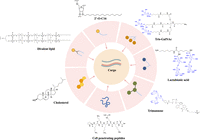
Ligand conjugated delivery strategies for ASO/siRNA delivery. Schematic depicting different cell/organ targeting ligands that are employed for ASO/siRNA delivery.
In addition to GalNAc, other liver-targeting carbohydrate ligands have also been explored. Lactobionic acid (LBA) has been shown to have similar efficacy to the GalNAc ligand for the delivery of PNAs with hepatocyte-enriched targeting (Kumar et al. 2023). This study compared divalent LBA with trivalent GalNAc in a series of in vitro and in vivo biodistribution and efficacy studies targeting miR-122. The LBA ligand also preferentially targets ASGPR to enter cells, similar to GalNAc. In vivo, studies indicated that diLBA-conjugated PNA targeting ASGPR had a 20-fold higher accumulation in the liver than unconjugated PNA.
Other sugars, such as mannose, have been explored for tissue-specific targeting of ASOs as alternatives to galactose-based ligands. Mannose receptors have been found on the surface of alveolar macrophages, which facilitate mannose ligands to target the lungs. To target inflammation in the lungs caused by COVID-19, an LNA targeting miR-21 conjugated to trimannose ligand showed better lung targeting efficacy than unconjugated LNA (Beck et al. 2023). The trimannose ligand targets the alveolar macrophages via the mannose receptor 1 (MRC1). The trimannose-conjugated miR-21 inhibitor targeted the macrophages in the lung, indicating the selective delivery of ASOs when delivered through inhalation. miR-21 upregulation was reversed with carbohydrate-conjugated miR-21 inhibitor compared with unconjugated nucleic acid. In another study, trimannose was conjugated to siRNA to target the pancreatic macrophages through the MRC1 receptor (Yamazaki et al. 2023). Biodistribution studies revealed the preferential uptake of trimannose-conjugated siRNA in the pancreas, liver, and kidney. Overall, the versatility of carbohydrate ligands makes them an effective modality for ASO-targeted delivery.
CONCLUSIONS AND FUTURE DIRECTIONS
Ligand-conjugated nucleic acids can be used in a wide range of therapeutic applications. Receptors such as ASGPR and MRC1 have been targeted for tissue-specific delivery using carbohydrate-based ligands. Despite these successes, there is a need to develop targeting modalities specific for targeting extra-hepatic tissues such as kidneys, lungs, heart, CNS, and pancreas for broader clinical application. Several bioconjugates are being investigated in clinical trials (Table 1). On the other hand, nanocarriers have also been developed for active targeting purposes. However, optimum ligand density, release kinetics, conjugation strategy, and the presence of spacer molecules between the ligand and nanocarrier are some of the challenges that need to be addressed for ligand-decorated nanocarriers (Mi et al. 2020). Recent advances in chemical modifications for nucleic acids have provided better ASO stability following systemic administration, reducing the over-reliance on nanocarriers for delivery. Though nanocarriers such as polymeric nanoparticles and LNPs have also been explored for ASO delivery, they still encounter significant challenges in scale-up and batch-to-batch variation (Liu and Meng 2021; Herdiana et al. 2022). Therefore, direct biocompatible ligand conjugation to ASOs and siRNAs is emerging as the efficient strategy for targeted delivery of nucleic acid-based therapeutics.
Past and current clinical trials for nucleic acid-based bioconjugates
One of the significant criteria in the engineering of targeting ligands revolves around determining target receptor expression in diseased tissue. Several databases, such as the Human Protein Atlas, Oncomine, IUPHAR/BPS, Uniprot, MPS Truc, and MemProtMD, can provide valuable insights regarding receptor protein structure, gene and protein expression profiles, and functional assays. Thus, ligand engineering can be optimized to target receptors having three- to fivefold higher expression in diseased tissues for improved targeting. Artificial intelligence-based methods can screen different receptors on specific cell types and effectively design ligands with a high affinity for different target receptors. This modeling approach would save resources and expedite the process by optimizing ligands for organ-enriched targeting.
In addition to coding RNAs, bioconjugates can target noncoding RNAs such as miRNAs, long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs) in different diseases. Thus, increasing the ligand-conjugated repertoire will also expand the targeting of diverse kinds of RNA for nucleic acids-based drug development. Overall, ligand conjugation to nucleic acids has proven to be an effective, safe, and scalable way to deliver RNA therapeutics for clinical applications.
ACKNOWLEDGMENTS
The authors thank the following funding sources: National Institutes of Health (NIH) R01 (1R01CA241194-01A1) grant to R.B. and F.J.S. and NIH R35 CA232105 to F.J.S. All figures were created using BioRender.com
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080273.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








