
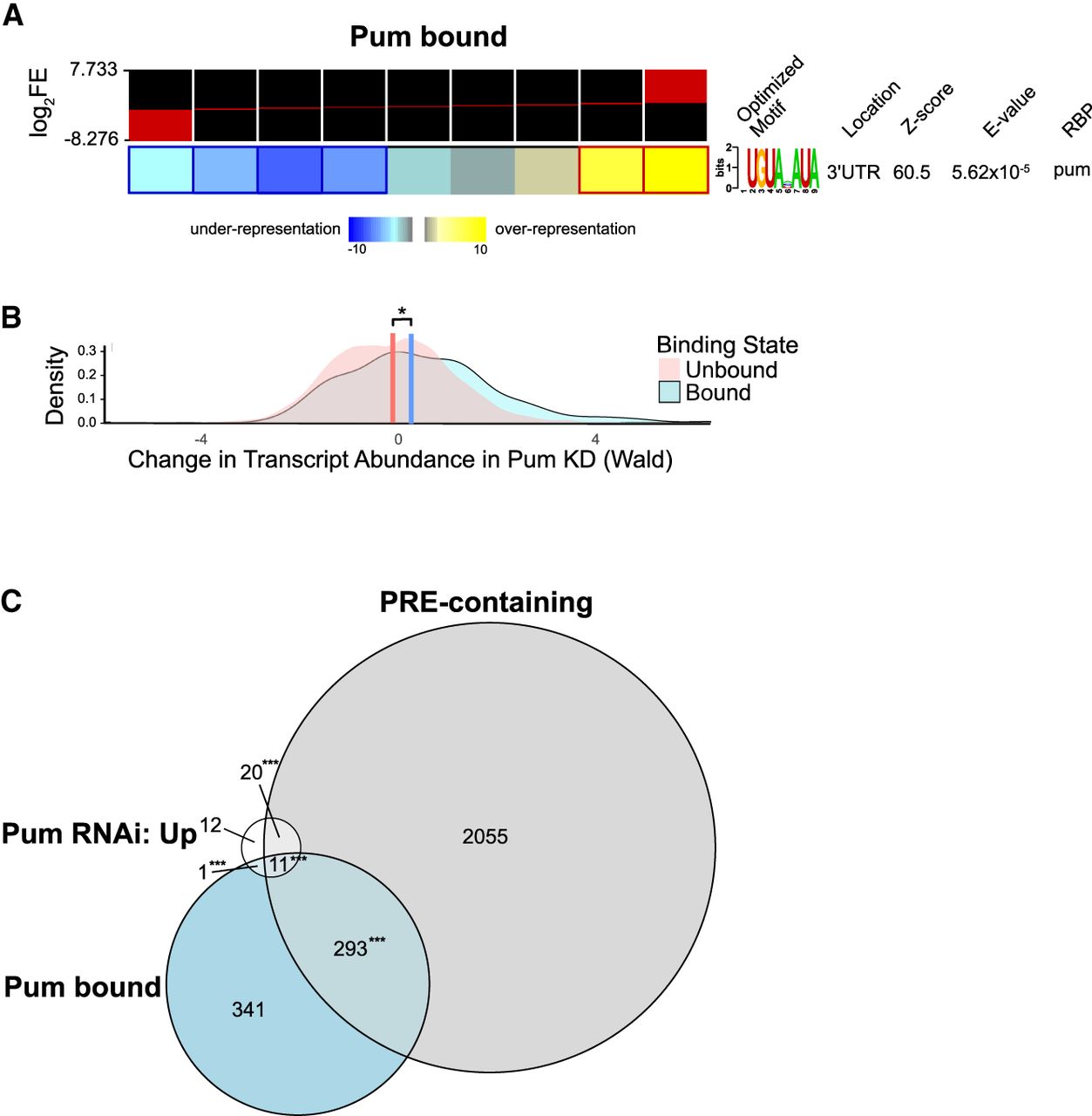
Analysis of Pum-bound mRNAs identifies PRE-containing, Pum-repressed direct targets. (A) Identification of a significantly overrepresented RNA sequence motif in Pum-enriched transcripts from Drosophila embryos identified by RIP-seq, analyzed using the FIRE algorithm. Target RNAs were sorted into nine equally populated bins, in order of log2 fold enrichment values in Pum RIP-seq data, as indicated at the top (the red sector in each bin indicates the range of log fold changes contained in that bin), and the over- or underrepresentation of the identified motif in each bin is shown with the blue–yellow color scale. Significant overrepresentation is observed for a motif that is highly identical to the documented Pum-binding site, the PRE, in the 3′ UTR of transcripts that are strongly enriched in the Pum RIP-seq. The Z-score output by FIRE indicates the information content of the optimized motif. The E-value output by TOMTOM indicates how confidently the motif matched with a known RBP. (B) Overlapping density distributions of the RNA abundance changes in response to Pum knockdown, plotted as significance-weighted log fold change (Wald statistic), for genes that were classified as bound (blue) or unbound (red) by Pum RIP-seq, based on a LFC ≥ 1.5 and a q-value ≤ 0.05. (C) Venn diagram comparing the overlap of significantly up-regulated genes by RNAi of Pum, measured by RNA-seq, with expressed PRE-containing genes and Pum-bound genes, identified by RIP-seq. Gene sets are reported in Supplemental Table S4. P-values for the significance of two-set or three-set gene overlaps were calculated via one-sided permutation tests (n = 1000), as described in the Materials and Methods section. For significance calling: (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.








