
RlmQ: a newly discovered rRNA modification enzyme bridging RNA modification and virulence traits in Staphylococcus aureus
- Roberto Bahena-Ceron1,
- Chloé Teixeira2,
- Jose R. Jaramillo Ponce1,
- Philippe Wolff1,
- Florence Couzon2,
- Pauline François2,
- Bruno P. Klaholz3,4,5,6,
- François Vandenesch2,7,8,
- Pascale Romby1,
- Karen Moreau2 and
- Stefano Marzi1
- 1Université de Strasbourg, CNRS, Architecture et Réactivité de l'ARN, 67000 Strasbourg, France
- 2CIRI, Centre International de Recherche en Infectiologie, Université de Lyon, Inserm U1111, Université Claude Bernard Lyon 1, CNRS UMR5308, ENS de Lyon, 69007 Lyon, France
- 3Centre for Integrative Biology, Department of Integrated Structural Biology, IGBMC, 67400 Illkirch, France
- 4CNRS UMR 7104, 67400 Illkirch, France
- 5Inserm U964, 67400 Illkirch, France
- 6Université de Strasbourg, 67000 Strasbourg, France
- 7Institut des agents infectieux, Hospices Civils de Lyon, 69004 Lyon, France
- 8Centre National de Référence des Staphylocoques, Hospices Civils de Lyon, 69317 Lyon, France
- Corresponding author: s.marzi{at}ibmc-cnrs.unistra.fr
-
Handling editor: Fatima Gebauer
Abstract
rRNA modifications play crucial roles in fine-tuning the delicate balance between translation speed and accuracy, yet the underlying mechanisms remain elusive. Comparative analyses of the rRNA modifications in taxonomically distant bacteria could help define their general, as well as species-specific, roles. In this study, we identified a new methyltransferase, RlmQ, in Staphylococcus aureus responsible for the Gram-positive specific m7G2601, which is not modified in Escherichia coli (G2574). We also demonstrate the absence of methylation on C1989, equivalent to E. coli C1962, which is methylated at position 5 by the Gram-negative specific RlmI methyltransferase, a paralog of RlmQ. Both modifications (S. aureus m7G2601 and E. coli m5C1962) are situated within the same tRNA accommodation corridor, hinting at a potential shared function in translation. Inactivation of S. aureus rlmQ causes the loss of methylation at G2601 and significantly impacts growth, cytotoxicity, and biofilm formation. These findings unravel the intricate connections between rRNA modifications, translation, and virulence in pathogenic Gram-positive bacteria.
Keywords
- m7G in 23S rRNA
- RNA modification
- methyltransferase
- tRNA accommodation
- Staphylococcus aureus virulence
INTRODUCTION
Ribosomal RNA (rRNA) is an essential component of the ribosome, the cellular machinery responsible for protein synthesis. It plays the most critical functional roles, assisting mRNA decoding, tRNA movements, and peptide bond formation (Nissen et al. 2000; Ogle et al. 2001; Demeshkina et al. 2012; Noller et al. 2017). Over the years, extensive studies have identified various post-transcriptional modifications that adorn rRNA molecules. These modifications, which are added during ribosome assembly (Siibak and Remme 2010), participate in the balance between translation speed and accuracy. Indeed, in vitro reconstituted Escherichia coli ribosomes lacking rRNA modifications were severely defective in catalytic activity (Green and Noller 1996), and the ribosome assembly was also altered (Cunningham et al. 1991). Numerous studies showed that the loss of rRNA modifications perturbs the active site structures in bacterial (Desaulniers et al. 2008; Demirci et al. 2010) and eukaryotic ribosomes (Natchiar et al. 2017), and causes altered rates and accuracy of translation (Liang et al. 2007).
In bacterial ribosomes, three primary types of rRNA modifications exist: pseudouridine (Ψ), methylation of the 2′-hydroxyl group of ribose (Nm), and methylation of bases (mN) (Decatur and Fournier 2002). While the precise roles of numerous modifications remain undetermined, they impart distinct characteristics to the nucleotides. For example, they can change the hydrogen bond capacities (Ψ) or decrease the stacking of bases (Dihydrouridine, D), impart structural rigidity (Ψ and Nm), or flexibility (mN) to both single- and double-stranded regions, potentially affecting hydrogen bonding (Arnez and Steitz 1994; Charette and Gray 2000). These modifications are concentrated in highly conserved and dynamic regions crucial for ribosomal functions including decoding, peptidyl transfer, the binding sites of A- and P-site tRNAs, the peptide exit tunnel, and the intersubunit bridges (Golubev et al. 2020; Watson et al. 2020).
The conservation of the position and chemical nature of the modified nucleotide across bacterial taxa has been a subject of growing interest. Several modifications appear to be strictly conserved, while others vary among bacteria. For instance, within the large ribosomal subunit of E. coli ribosome, Gm2251 and Um2552 are situated in the P- and A-loops, respectively, where they interact with the CCA-end of tRNAs in the P- and A-sites. While Gm2251 is conserved across the three domains of life (Sergeeva et al. 2015; Natchiar et al. 2017), Um2552, added by RlmE methyltransferase, is present in many bacteria, except for certain Bacillus species like Bacillus subtilis and Bacillus stearothermophilus. Instead, in these bacterial species, a different methyltransferase, RlmP, methylates G2553 during 50S biogenesis (Hansen et al. 2002; Roovers et al. 2022). In the rlmE-deficient E. coli mutant strain, the absence of Um2552 modification, which decreases the flexibility of adjacent nucleotides, leads to a delay in 50S subunit maturation, to a slower subunit association, and translocation rate (Wang et al. 2020). Translation with unmethylated U2552 in E. coli appears to be more accurate (Widerak et al. 2005) and to proceed at a reduced rate (Caldas et al. 2000). These results suggested that a certain level of recoding provided by the methylation can be accepted to favor higher translation speed important for cellular physiology. Numerous rRNA modifications have also been observed along the tRNA transition pathway, both on the 30S and the 50S subunits (Watson et al. 2020). During early ribosome assembly, the methyltransferase RlmI methylates position 5 of C1962 (Purta et al. 2008), which is located on helix H70 at the edge of its coaxial stacking with H71. Together with H69, which contains the conserved Ψ1911 (RluD), m3Ψ1915 (RluD, RlmH), and Ψ1917 (RluD), these rRNA helices provide a sliding support for the movements of tRNAs into the ribosome (Girodat et al. 2023). The rlmI depletion in a mutant E. coli strain led to a small growth defect in competition experiments (Purta et al. 2008) or when translation is enhanced following the overexpression of an induced gene (Pletnev et al. 2020).
In the present study, we have identified a paralog of E. coli RlmI in Staphylococcus aureus. Unexpectedly, we find that the enzyme is responsible in vivo for the methylation at position 7 of G2601 located in helix H90 of the 23S rRNA. Importantly, this position is unmodified in E. coli (G2574) hinting at a specific function in S. aureus. Because we did not detect any methylation at C1989 of S. aureus 23S rRNA (equivalent to E. coli C1962 methylated at position 5), we have renamed this new methylase, RlmQ. Phenotypic analysis of the rlmQ-disruption mutant strain revealed a slightly slower doubling time in nutrient-poor media, a reduced cytotoxicity, and an increased biofilm production. In the following, we will also discuss the evolutionary considerations of RlmQ and RlmI, two highly similar enzymes with distinct specificities for 23S methylation in distant bacterial species, and their biological consequences.
RESULTS AND DISCUSSION
S. aureus 23S rRNA contains a 7-methylguanosine at position 2601
Given the important roles of RNA modifications on bacterial physiology, we wondered whether the Gram-positive S. aureus bacteria harbor different sets of RNA modifications compared to the evolutionary distant Gram-negative bacteria E. coli. Here, we have focused the study on the modifications present in S. aureus 23S rRNA. After purification of the S. aureus 23S rRNA from the HG001 strain and isolation of specific fragments by RNase H cleavage, the identification of RNA modifications was done using RNase T1 digest followed by LC/MSMS analysis. Among the various sequenced oligoribonucleotides, a fragment with a mass of 1948.2 Da (m/z 973.62) corresponded to the UAC[mG]CG > p sequence. Subsequent MSMS fragmentation revealed the presence of a methylation at G2601 located in H90 (corresponding to E. coli G2574; Fig. 1A,B). To visualize the presence of the methylation and its position on the guanosine, we conducted an in-depth analysis of the high-resolution structure of S. aureus ribosome (Halfon et al. 2019) (map: EMD-10077; PDB: 6s0z; Fig. 1C). Within this map, we identified a density that extends into position 7 within the purine ring, in favor of the presence of a 7-methylguanosine (m7G), not modeled. Finally, the presence of m7G was validated by primer extension (PE) analysis, adapting a specialized protocol originally designed for genome-wide mapping of m7G (Marchand et al. 2018). The experiments showed a strong band corresponding to a specific termination of reverse transcription (RT) due to the m7G2601 in both the virulence-attenuated HG001 and the virulent USA300 (LAC) strains (Fig. 1D). Interestingly, this modified position has also been reported in B. subtilis (Hansen et al. 2002) suggesting that m7G2601 in the helix H90 is conserved among firmicutes. E. coli ribosome structure suggests that H90 serves as a corridor to facilitate the accommodation of the aminoacylated CCA-end of tRNA (aa-CCA) into the A-site of the peptidyl transferase center (PTC) (Fig. 1E; Whitford et al. 2010). Although the resolution of the cryo-EM structure of S. aureus 70S ribosome is lower than E. coli 70S and no A-site tRNA is present (Golubev et al. 2020), the positioning and structures of H90 and neighbor rRNA helices are highly similar. The accommodation process involves key conformational changes in both tRNA and ribosome that significantly impact the rate of cognate tRNA selection (Rodnina and Wintermeyer 2001). A kinetic study combined with dynamic simulation has revealed the relatively slow nature of this process due to spontaneous and reversible accommodation attempts, akin to a stochastic trial-and-error mechanism, encompassing various parallel pathways (Whitford et al. 2010). Notably, the corridor traversed by the aa-CCA of the tRNA is nearby m7G2601 in S. aureus. Hence, it is tempting to propose that this methylation might be one of the key players which facilitates the accommodation process. More experiments will be required to assess the impact of the m7G modification on translation dynamics, especially its selectivity and functional importance for different aminoacyl-tRNAs.
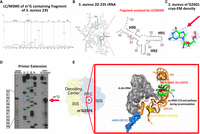
S. aureus ribosome contains m7G2601. (A) Mass spectrometry analysis of the RNase T1 derived fragment of S. aureus HG001 23S rRNA region spanning from U2598 to G2603 (corresponding to E. coli U2571 to G2576). The fragment has a mass of 1948.2 Da (m/z 973.62) and carries the sequence UAC[mG]CG > p based on MSMS analysis. (B) 2D representation of the entire S. aureus 23S rRNA, zooming on the region containing helix H90 with G2601 (corresponding to E. coli G2574). Red and black numbers indicate S. aureus and E. coli numbering, respectively. The red bar highlights the fragment analyzed by LC/MSMS. (C) Structural analysis of G2601 from the deposited S. aureus 50S cryo-EM map at 2.3 Å (Halfon et al. 2019) (map: EMD-10077; PDB: 6s0z). The density around N7 indicates the presence of the methyl group, which is not modeled in the atomic coordinates. (D) PE analysis of HG001 and USA300 (LAC) S. aureus strains, including controls for the AlkAlanine treatment. AGCU lanes represent dideoxy sequencing, NT stands for the nontreated sample, and the red arrow points to the reverse transcriptase stop due to the m7G-modified nucleotide. (E) Details of the accommodation corridor for aminoacyl-tRNA CCA-end on the E. coli ribosome structure, with the A-site tRNA (gray surface) in the accommodated state (Watson et al. 2020). The E. coli 23S rRNA helices (H69–H71, H90–H91) are shown: G2524 (red sphere), corresponding to m7G2601 in S. aureus, is located in H90 (green), while m5C1962 (yellow sphere), corresponding to C1989 in S. aureus, is in H70/H71 (bright orange). The mRNA (blue transparent surface) in the 30S decoding channel (DC) interacts with the anticodon of the A-site tRNA. (Sau) S. aureus, (Eco) E. coli.
In quest of the enzyme responsible for m7G2601
To identify the enzyme responsible for m7G2601 in S. aureus, we have first performed a bioinformatic analysis, primarily focusing on sequence similarities through BlastP and sequence alignments. Our approach has involved three representative methyltransferases from E. coli, S. aureus, and B. subtilis, which are known for their methylation activity on 16S rRNA (RsmG for m7G967—E. coli numbering), 23S rRNA (the carboxy-terminal half of E. coli RlmKL corresponding to B. subtilis RlmK, for m7G2069—E. coli numbering), and tRNA (TrmB for m7G46 in the variable loop of different tRNAs). We utilized BlastP with the sequences of the catalytic S-adenosyl-methionine (AdoMet) binding domains (MTase) from the selected m7G methylases to search for similar proteins in S. aureus NCTC8325, which is highly similar to the HG001 strain. The analysis revealed many members of the COG1092 methyltransferase family, encompassing m5C MTases and m7G MTases, due to the high similarity of their catalytic domains. Among the identified candidates in S. aureus, only seven are predicted enzymes with unknown functions. To ascertain whether one of these enzymes was responsible for the presence of m7G2601, we systematically examined the PE profile of 23S rRNA prepared from the mutant strains carrying individual gene disruption for each candidate (Fig. 2A). These strains were selected from the Nebraska Transposon Mutant Library collection and compared with the isogenic wild-type (WT) USA300 JE2 strain (Fey et al. 2013). The PE analysis showed that only the mutation at the locus SAUSA300_0981 in USA300 (HG001_00931 in HG001 strain) causes the loss of the specific RT pause at G2601.
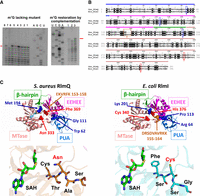
RlmQ is a m7G methyltransferase responsible for the enzymatic modification of m7G2601. (A) AlkAniline PE analysis of S. aureus 23S rRNA from USA300 JE2 WT and seven selected strains from the Nebraska collection with disrupted genes for methyltransferases of unknown function. AGCU dideoxy sequencing lanes. (Left) Lane 1, USA300 WT; lane 2, NE553; lane 3, NE1671; lane 4, NE1442; lane 5, NE189; lane 6, NE807; lane 7, NE1546 (corresponding to rlmQ-disruption mutant); lane 8, NE1337. (Right) Analysis of m7G restoration in the complemented mutant strain. Lane 1, S. aureus USA300 Lac WT-pCN38:wP:TT; lane 2, NE1546-pCN38:wP:TT; lane 3, NEB1546-pCN38:wP:rlmQ:TT. Red arrows indicate m7G modification. (B) Sequence alignment of S. aureus RlmQ and E. coli RlmI. Distinct domains are highlighted with colored bars as follows, starting from the amino terminus: PUA domain (blue bar over the sequence), EEHEE domain (magenta bar), β-hairpin (green bar), and MTase domain (red bar). Colored boxes spotlight the most significative signatures discriminating RlmQ and RlmI enzymes, as determined by BIS2 analysis (Dib and Carbone 2012). Red boxes represent cluster 1, with a P-value cutoff at 4.3 × 10−6, revealing the substitution of the conserved catalytic cysteine, typically found in m5C methyltransferases, by asparagine in RlmQ. Within this cluster, another substitution (His/Phe) correlates with the former, contributing to the distinctive motifs for the two classes of methyltransferases. Blue boxes denote cluster 2, with a P-value cutoff at 2.6 × 10−5, encompassing Lys/Met, Pro/Gly, and Arg/Trp covariations in RlmI/RlmQ, respectively. The brown box indicates cluster 3, with a P-value cutoff at 3.7 × 10−5, associating the fragment sequences DRSDVAVRKK/EKVRFK (RlmI/RlmQ) with the covariations of cluster 1. (C) Structure analysis illustrating the specific covariation clusters characterizing RlmQ (on the right) and RlmI (on the left). The structures of RlmQ and RlmI are sourced from the pdb 3VSE (Kita et al. 2013) and pdb 3C0K, respectively. The domains and motifs in the figure follow the same color code as in B. At the bottom, a comparison of the MTase active sites is presented for S. aureus RlmQ (orange background structure) and E. coli RlmI (cyan background structure). In the motif FSCSG, E. coli RlmI contains a cysteine (Cys), whereas S. aureus RlmQ has an asparagine (Asn) in the motif CTNAS. To facilitate comparison, the substrate S-adenosyl-l-homocysteine (SAH), present only in the RlmQ crystal structure, has also been modeled in the RlmI structure.
To ensure that the differences observed were solely attributable to SAUSA300_0981 disruption, we sequenced and analyzed the genomes of both WT and the mutant strains. The presence of the transposon at position 451 of the SAUSA300_0981 gene sequence (1173 pb long) was confirmed and no other sequence change in the genome was detected. At the point of insertion, a stop codon in the frame with the coding region was generated (Supplemental Fig. 1) preventing the translation of full-length protein. Through RT-PCR, we demonstrated that the transposon insertion led to the synthesis of a shorter transcript, lacking the sequence encoding the catalytic MTase domain (Supplemental Fig. 1). This strongly suggests that the mutation cannot lead to the synthesis of an active enzyme. Subsequently, we conducted a complementation of the mutant strain by transforming it with a plasmid expressing the WT SAUSA300_0981 gene under the control of a constitutive mild promoter. A mass spectrometry analysis of total proteins from the WT, the mutant strain, and the complemented strain confirmed the absence of spectra for the SAUSA300_0981 protein in the mutant. In contrast, several spectra corresponding to different regions of the protein were observed in the WT and complemented strains (Supplemental Fig. 1).
The PE analysis showed that the premature RT stop was restored in the complemented strain, as shown for the WT JE2 strain (Fig. 2A). All in all, these experiments show that SAUSA300_0981 encodes the methyltransferase responsible for the 7-methylation of G2601. In accordance with the established bacterial nomenclature for rRNA methyltransferases, we have named this newly identified methyltransferase “RlmQ.”
S. aureus RlmQ and E. coli RlmI methyltransferases are paralogues with distinct activities
Based on its amino acid sequence, HG001_00931 (or SAUSA300_0981 in USA300) gene locus was previously annotated as RlmI (Caldelari et al. 2017), the RNA methyltransferase responsible for m5C1962 in 23S rRNA from E. coli (corresponding to S. aureus C1989) and various bacteria (Purta et al. 2008). Indeed, they share 46% sequence similarity and signatures for similar domain organization (Fig. 2B). To perform a more detailed analysis of the similarities and differences between these two 23S rRNA methyltransferases, we conducted an extensive search in the PDB database using RlmQ as the reference (Holm et al. 2023). Remarkably, we identified the crystal structure of an S. aureus methyltransferase named SAV1081 (Kita et al. 2013), with no attributed function but which displayed 100% coverage and sequence similarity to RlmQ. This structure displays three main domains: a Rossman-Fold methyltransferase domain (MTase), the methyltransferase conserved RNA recognition module EEHEE domain, and the RNA binding domain PUA, which is responsible for the recognition of specific RNA sequences (Fig. 2C; Sunita et al. 2008). The EEHEE and MTase domains are interconnected by a conserved β-hairpin structure, a feature shared with several other methyltransferases. Superposition of E. coli RlmI and S. aureus RlmQ structures (pdb files 3VSE for RlmQ and 3C0K for RlmI) shows a root mean square deviation (RMSD) of 1.45 Å and a TM-score of 0.4254 (Zhang and Skolnick 2004), indicating a strong overall structural similarity. Both enzymes belong to the COG1092 family, featuring a well-conserved AdoMet binding site in the MTase domain. This domain shares 28.5% of sequence identity, 45.2% of sequence similarity, and a TM-score of 0.2518 between the two enzymes. Additionally, their EEHEE domains exhibit a high degree of conservation with 32.7% sequence identity and 54.8% sequence similarity. Structure comparison also reveals similarity (TM-score = 0.321). In contrast, their amino-terminal PUA domains display the most pronounced sequence and structural divergence with 21.3% sequence identity and 41.3% sequence similarity. The TM-score for this region is 0.2518. Within the MTase active site, the principal distinction arises from a specific amino acid substitution: a catalytic cysteine (C) in RlmI is substituted by an asparagine (N) in RlmQ (Fig. 2C). The presence of this cysteine in eubacteria, archaea, and certain plants (Kita et al. 2013) confers m5C RNA methyl transferase activity (Liu and Santi 2000; King and Redman 2002), while the presence of an asparagine has been found in a separate subgroup of the COG1092 family in methylases in several Firmicute members (Sunita et al. 2008). To find sequence and structural motifs characteristic of RlmQ and RlmI enzymes, we searched for coevolution signals among pairs of amino acids or protein sequence fragments using the BIS2 software (Dib and Carbone 2012). We initially obtained homologous sequences from distinct bacterial taxa for RlmQ and RlmI independently. This retrieval was performed from the manually annotated section of UniprotKB using psiblast (Altschul et al. 1997) at the website (https://www.ebi.ac.uk/Tools/sss/psiblast/) with default setting. We subsequently conducted a multiple sequence alignment (MSA) on a representative collection of 19 homologous protein sequences (Supplemental Fig. 2A) taken from different taxa and used it for BIS2 analysis. The analysis, allowing a maximum of one exception among the sequences, has identified different coevolution clusters which have been mapped on the sequence alignment (Fig. 2B; Supplemental Fig. 2A), and on RlmQ and RlmI structures (Fig. 2C). The most significant cluster (cluster 1, with a P-value cutoff at 4.3 × 10−6) provides additional evidence for the species-specificity of Asn (RlmQ) and Cys (RlmI) residues, together with their coevolved cluster partners, the structurally close Phe (RlmQ) and His (RlmI) residues within their MTase domain. In addition, a slight reduction in the significance cutoff (P-value cutoff at 3.7 × 10−5) revealed correlated variations in the PUA, EEHEE, and β-hairpin domains as well (Fig. 2B,C; Supplemental Fig. 2A). Our analysis also revealed two intriguing exceptions. In the γ-proteobacterium Pseudomonas aeruginosa, both RlmQ and RlmI modification enzymes coexist. Whether both modifications (m5C and m7G) could occur on the same 23S rRNA remained to be demonstrated. Among the Firmicute phylum, Clostridium perfringens has a methylase enzyme carrying all the peculiarities of the RlmI class.
To investigate the potential involvement of RlmQ in the modification of m5C1989 (E. coli C1962), we conducted a meticulous analysis using mass spectrometry on the relevant 23S rRNA region from S. aureus. We then compared it with the corresponding region of E. coli 23S rRNA, where modification m5C1962 by RlmI was anticipated (Fig. 3A). The 23S rRNA, isolated from S. aureus HG001 strain and E. coli MRE600 strain, was first hybridized to two specific oligonucleotides and digested with RNase H. This treatment led to specific RNA fragments of 25 nt, which include rRNA helices H70 and H71 and the expected target cytosine (Fig. 3B). After gel electrophoresis purification, the gel bands containing the 25 nt long RNA fragments were further treated with RNase T1. Subsequent LC/MSMS analysis of S. aureus sample revealed the presence of a short fragment with a mass of 1606.21 Da corresponding to the unmodified sequence ACCCGp (Fig. 3A, upper panel). In contrast, for E. coli, we detected a fragment of 1621.2 Da corresponding to the modified sequence AC[m5C]UGp (Fig. 3A, lower panel). Comparison of the density maps from the reported high-resolution structures of E. coli and S. aureus ribosomes reveals clearly the presence of a methyl group at position 5 of C1962 in E. coli 23S rRNA, while this methyl is likely absent at position 5 of the equivalent C1989 in S. aureus (Fig. 3C).
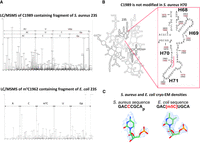
RlmQ/RlmI evolution changes 23S methylation maps in S. aureus and E. coli. (A) Detection of nonmodified C1989 in S. aureus (upper panel) and m5C1962 in E. coli (lower panel) using mass spectrometry. The RNase H generated fragment including H70 and H71 of the 23S rRNAs has been further digested with RNase T1 before analysis. S. aureus MSMS spectrum (upper panel) contains three fragments including the sequence of interest ACCCGp showing no methylation. E. coli MSMS spectrum (lower panel) contains only the methylated fragment of interest AC[m5C]UGp. (B) 2D representation of the entire S. aureus 23S rRNA, zooming on the region containing H70 and H71, with C1962 (E. coli numbering, corresponding to S. aureus C1989). Red and black numbers indicate S. aureus and E. coli numbering, respectively. The red bar highlights the fragment analyzed by LC/MSMS. (C) Structure analysis of C1962 from the deposited S. aureus 50S cryo-EM map at 2.3 Å (Halfon et al. 2019) (map: EMD-10077; PDB: 6s0z) and E. coli deposited 70S cryo-EM map at 2 Å (Watson et al. 2020) (map: EMD-22586; PDB: 7k00). While a clear density for methylation of position 5 on C1962 is observed in E. coli 23S allowing building this modification in the fitted model, no extra density is present on S. aureus 23S.
Taken together, we show in vivo that S. aureus RlmQ methylates the N7 position of G2601 (equivalent to G2574 in E. coli), while C1989 is not modified (equivalent to C1962 in E. coli). Because this methylation has also been observed in B. subtilis, we propose that Firmicutes share the same features. Conversely, in Gram-negative bacteria, RlmI is the enzyme which methylates C1962 at its position 5, while no existing modification was attributed to G2574. It is conceivable that these two paralogous enzymes may have originated from a gene duplication event and divergent evolution in these distant species (Supplemental Fig. 2B). Interestingly, E. coli m5C1962 and S. aureus m7G2601 are both situated along the pathway taken by tRNAs during their transit into the ribosome (Fig. 1E; Watson et al. 2020). In this scenario, the tRNAs do not make direct contact with helices H70 and H71, although these helices are in close proximity to the tRNA acceptor arms and variable loops. It is plausible that Gram-negative and Gram-positive ribosomes have evolved slightly different solutions to facilitate the smooth transition of tRNAs within the ribosome, both involving rRNA methylation, albeit targeting distinct residues.
Phenotypic characterization of S. aureus rlmQ mutant
S. aureus is an opportunistic pathogen, which has evolved many strategies to regulate the synthesis of numerous virulence factors in response to the host, stress, and various environmental changes. To assess the impact of RlmQ enzyme modification on the physiology of S. aureus, the WT USA300 JE2 strain was compared to a mutant deficient for the rlmQ enzyme (rlmQ−) in various phenotypic tests including growth rate, biofilm, and cytotoxicity on human cells (Fig. 4).
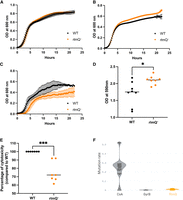
Phenotypic characterization of S. aureus rlmQ mutant. (A–C) Growth of the WT strain and the mutant inactivated for rlmQ (rlmQ−) was compared under different media and temperature conditions. Bacterial growth was measured during 24 h in BHI medium at 37°C (A), 42°C (B), and in RPMI medium at 37°C (C), n = 2–6. (D) Biofilm was quantified by crystal violet staining of the extracellular matrix after 24 h of culture in BHI medium, n = 8. (E) Bacterial supernatant cytotoxicity was quantified by measuring propidium iodide (PI) incorporation into C5aR1-transfected U937 monocytes, n = 7. Mann–Whitney tests performed: (*) P-value < 0.05, (**) P-value < 0.01, (***) P-value < 0.001. (F) Mutation rate of protein sequences from 301 clinical strains was estimated with a ratio: number of missense mutations divided by sequence match length.
We first analyzed whether the mutation had an effect on S. aureus growth. Under optimal growth conditions (BHI medium, at 30°C and 37°C), no difference was observed between the WT and the rlmQ− mutant strains (Fig. 4A; Supplemental Fig. 3). This is in agreement with the fact that the polysome profiling in the WT and mutant strains remained identical in the two strains under these conditions of growth (Supplemental Fig. 4). These experiments also show that the maturation process of the ribosome is not significantly affected by the mutation. Only at 42°C in BHI, the rlmQ− mutant reached a slightly higher OD at the stationary phase than the WT strain (Fig. 4B). The two strains were cultured in a less rich environment (RPMI medium) closer to the conditions encountered during infection. Under these conditions, we observed a significant reduction in the growth of the mutant strain compared to the WT strain with a reduced doubling time at the exponential phase (rlmQ− 65 min/WT 45 min), and a diminution of the maximal OD (rlmQ− 0.38/WT 0.56) (Fig. 4C). We then monitored the ability of S. aureus WT and mutant strains to form biofilm which play major roles in an infectious context. The amount of biofilm formed on plates by the rlmQ− mutant was significantly greater than that of the WT strain (Fig. 4D). Another important feature of bacterial–host interaction in infectious conditions is the cytotoxic activity of S. aureus against immune cells. The cytotoxic activity of the WT and mutant strains was assessed on human monocytes expressing the C5aR1 receptor targeted by two pore-forming toxins, the Panton–Valentine leucocidin (PVL) and the γ toxin CB (HlgCB). Monocytes were exposed to overnight culture supernatants (which contain bacterial toxins but no live bacteria). The cytotoxic activity of these supernatants was assessed by the incorporation of PI into the DNA of cells with altered plasma membranes. The rlmQ− mutant showed a significant reduction in cytotoxic activity compared to the WT strain (Fig. 4E).
We finally addressed whether RlmQ is conserved among clinical infection isolates. To this end, the rlmQ gene was searched in the genomes of a collection of 301 well-characterized clinical S. aureus strains from previously published cohorts. The data showed that the rlmQ gene was detected in all 301 strains and had the same size in 298 strains (Fig. 4F). Furthermore, the protein sequence was extremely well conserved with a mutation rate of 0.008 comparable to the highly conserved GyrB (MR = 0.002) but much lower than that of coagulase (MR = 0.263), which is known to be highly variable among strains. Using a Random Forest model based on horizontal oligonucleotide frequency, a conserved promoter sequence was predicted between −432 and −475 nt upstream of the initiator codon for the 301 strains (Supplemental Fig. 5).
Taken together, our data suggest that the highly conserved RlmQ methylase is playing an important role in human infections. Slower growth and increased biofilm formation are indicative of a chronic infection mode, and conversely, toxin production is a marker of acute infection. As RlmQ is associated with a better growth under physiological conditions, with an increase in cytotoxic activity, and a decrease in biofilm formation, we suggest that RlmQ might be important for the establishment of acute infection.
Conclusion
We showed here that S. aureus RlmQ methylase, paralogous to E. coli RlmI, is the enzyme which methylates the position 7 of G2601 located in the helix H90 of 23S rRNA. Interestingly, the two paralogous proteins have evolved different activities in these taxonomically distant bacterial species, which show different modification patterns (in particular, G2574 in E. coli is not modified). Although the two enzymes act on two different modification sites, their target nucleotides are located in a large structured region of the 50S subunit, which was proposed to accommodate the transition of tRNAs during translation. Whether this divergent evolution between Gram-positive and Gram-negative bacteria may have consequences on ribosome functioning remained to be studied. Interestingly enough, we demonstrated that under conditions closer to infectious conditions, the rlmQ mutant strain displays clearly distinct phenotypes compared to the WT strain, suggesting that RlmQ could play an important role during infection and/or colonization of the human host. Further experiments will be required to demonstrate if this is the modification of the 23S rRNA which contributes to these phenotypes or if RlmQ may have additional functions and RNA substrates in S. aureus. This study highlights that the functions of rRNA-specific modification enzymes expand to adaptive responses such as stress, environmental changes, and during infections caused by pathogenic bacteria, a domain that needs to be explored.
MATERIALS AND METHODS
Bacteria strains, plasmids, and growth conditions
The selected transposon mutants (NE553, NE1671, NE1442, NE189, NE807, NE1546, and NE1337) were obtained from the Nebraska mutant library produced by transposon bursa aurealis insertion (Fey et al. 2013). We also used 301 clinical strains isolated from patients suffering from three different types of infection as follows: 126 strains responsible for bacteremia with or without endocarditis from the French national prospective multicenter cohort VIRSTA ([Le Moing et al. 2015], ENA accession numbers: PRJEB49354, PRJEB48298), 166 strains from the French severe community-acquired pneumonia cohort ([Gillet et al. 2021], ENA accession number: PRJEB54685), and nine strains from necrotizing soft tissue infections (Baude et al. 2019).
Glycerol stocks from S. aureus USA300 LAC (Los Angeles County strain), a community-acquired methicillin-resistant isolate of the USA300 lineage, USA300 JE2, which lacks two plasmids compared to USA300 LAC (Fey et al. 2013), HG001, a derivative of the RN1 (NCT8325) strain with restored rbsU (Herbert et al. 2010; Caldelari et al. 2017), and the selected transposon mutants (NE553, NE1671, NE1442, NE189, NE807, NE1546, and NE1337) were all streaked in BHI agar and growth overnight at 37°C. Starter cultures were prepared the following day by inoculating isolated colonies in 2 mL BHI medium and incubating them at 37°C with agitation at 180 rpm for 16 h. Overnight cultures were diluted to OD600 nm 0.05 in 20 mL BHI and grown to late exponential phase at 37°C. Cells were harvested by centrifugation at 2100g for 10 min at 4°C and stored at −20°C. Chloramphenicol (10 µg/mL) was added to the medium when appropriate. Strains were also cultured in BHI broth at 37°C overnight, diluted at OD600 nm = 0.05 in fresh medium (BHI or RPMI). A 96-well plate was seeded with 200 mL of BHI and incubated at 37°C, 30°C, or 42°C in a Tecan plate reader for 24 h. OD at 600 nm was measured every 15 min after agitation.
Complementation of the rlmQ mutant strain (NE1546) was achieved by introducing the plasmid pCN38:wP:rlmQ:TT containing the rlmQ gene under a weak constitutive blaZ-derived promoter. Sequences of promoter and primers used for cloning are shown in Supplemental Table S1. The coding DNA sequence (CDS) and Shine–Dalgarno region of rlmQ were PCR amplified from S. aureus HG001 genomic DNA (HG001 and USA300 rlmQ CDS are identical) using primers rlmQ-F and rlmQ-R and cloned in the vector pEW (Menendez-Gil et al. 2020) between a modified blaZ promoter (wP) and the native blaZ transcriptional terminator (TT). Primers PblaZ-F and TT-rev were then used to amplify the region wP:rlmQ:TT from pEW:rlmQ, and the resulting fragment was subcloned in vector pCN38 (Charpentier et al. 2004). Plasmid pCN38:wP:rlmQ:TT was electroporated into S. aureus RN4220 strain before being introduced in NE1546 strain as described elsewhere (Schneewind and Missiakas 2014), and transformants were selected with chloramphenicol. For complementation assays, the empty pCN38 plasmid was introduced in WT USA300 JE2 and NE1546 strains.
RNA extraction and purification of 23S ribosomal RNA
Total RNA from S. aureus was prepared as previously described (Antoine et al. 2019). 23S rRNA was purified from total RNA by size-exclusion chromatography using a Superose 6 10/300 column (GE Healthcare) eluted at 0.4 mL/min with 300 mM ammonium acetate. Fractions were analyzed in 1% agarose gel in TBE buffer and those containing pure 23S rRNA were pooled and concentrated by ethanol precipitation.
AlkAniline treatment and primer extension
Detection of m7G was performed by AlkAniline treatment as described in Marchand et al. (2018). Briefly, 5 µg of total RNA in 10 µL of water was mixed with 10 µL of 100 mM NaHCO3 (pH 9.2) and incubated at 96°C for 5 min. Alkaline hydrolysis was stopped by precipitating the RNA with 10 µL 3M sodium acetate (pH 5.2), 1 µL glycoblue, and 1 mL of ethanol overnight at −20°C. Treated RNA was pelleted by centrifugation (16,000g for 15 min at 4°C), washed with 70% ethanol, and dried under vacuum in a SpeedVac (Savant). RNA pellet was resuspended in 20 µL 1 M aniline (pH 4.2) and incubated at 60°C for 15 min. Aniline cleavage was stopped by precipitating RNA as above.
Analysis of 23S rRNA G2575 position in treated RNA was performed by PE using DNA primer H90 complementary to region 2645–2666 (Supplemental Table S1). Ten picomoles of primer were 32P-5′-radiolabeled by incubation with γ-[32] ATP and 10 units of T4 polynucleotide kinase for 1 h at 37°C. RT reactions were performed in a final volume of 10 µL. First, 100,000 cpm of labeled primer were incubated with 1 µg of treated RNA at 80°C for 90 sec, then placed in ice for 90 sec to allow hybridization. Two microliters of RT buffer 5× were added, and the samples were incubated at 20°C for 15 min before the addition of 1.5 µL of dNTP mix (1 mM of dATP, dGTP, dCTP, dUTP) and 1 µL of AMV enzyme (PROMEGA). RT was performed at 42°C for 45 min, and synthesized cDNA was recovered by ethanol precipitation as described above. Dried pellets were resuspended in 6 µL urea-blue buffer (8 M urea, 0.025% xylene cyanol, and bromophenol blue), and 30,000 cpm of each sample were separated by electrophoresis at 75 W in a denaturing 10% polyacrylamide–7 M urea gel until the bromophenol blue reached the bottom. The gel was then placed inside a cassette with an autoradiography (Kodak) and incubated overnight at −80°C.
Mass spectrometry
LC/MSMS analysis was performed as previously described (Antoine et al. 2019). Modification at m7G2601 was identified by in-gel RNase T1 digestion of a specific S. aureus 23S rRNA fragment (nucleotides 2521–2617) isolated by RNase H cleavage using DNA primers H90-1 and H90-2 (Supplemental Table S1). The lack of methylation at C1989 in S. aureus and the presence of m5C1962 in E. coli was validated by in-gel RNase T1 digestion of specific 23S rRNA fragments obtained by RNase H cleavage using DNA primers H70-1 and H70-2 (nucleotides 1938–2090), and H70-1 and H70-3 (nucleotides 1912–2063), respectively (Supplemental Table S1). RNase H reaction was performed in a final volume of 10 µL. First, 5 µg of pure 23S rRNA were incubated with 10 µg of primers at 80°C for 2 min and slowly cooled down to 50°C to allow hybridization. Then, 0.5 U of RNase H (Thermo Fisher Scientific) in the appropriate commercial buffer was added, and digestion was for 30 min at 50°C. The RNase H fragment was isolated in a denaturing 10% polyacrylamide–urea gel and excised under UV light. RNase T1 digestion (20 µL) was performed with 20 U of enzyme in 100 mM ammonium acetate pH 6.8 at 50°C for 4 h. Samples were desalted using ZipTip C18 (Millipore) by several washes with 200 mM ammonium acetate, eluted in 50% acetonitrile and dried under vacuum. The pellet containing the RNase digest was resuspended in 3 µL of milli Q water and separated on a nanoAcquity UPLC system (Waters) equipped with an Acquity UPLC peptide BEH C18 column (130 Å, 1.7 µm, 75 µm × 200) equilibrated at 300 nL/min, with a buffer containing 7.5 mM triethylammonium acetate, 7 mM triethylamine, and 200 mM hexafluoroisopropanol. After loading, the oligoribonucleotides were eluted first with a gradient of methanol (15%–35%) for 2 min, followed by another gradient of methanol (35%–50%) for 20 min. Mass spectrometry (MS) and MS/MS analysis were performed using a SYNAPT G2-S instrument (Waters) equipped with a NanoLockSpray-ESI source in negative mode with a capillary voltage set to 2.6 kV and sample cone of 30 V. The source was heated to 130°C. Samples were analyzed over an m/z range from 500 to 1500 for the full scan, followed by a fast data direct acquisition scan (Fast DDA). Collision-induced dissociation (CID) spectra were deconvoluted using MassLynx software (Waters) and manually sequenced by following the y and/or c series.
Polysome profiling
S. aureus USA300 and NE1546 strains were cultivated in 20 mL BHI medium at 37°C until OD600 nm 2.0. Cells were resuspended in 500 µL lysis buffer (20 mM Tris pH 8, 100 mM NH4Cl, 50 mM MgCl2, 0.4% Triton X-100, 0.1% nonidet P-40), transferred into a tube with lysis matrix B (MP Biomedicals), and lyzed by bead beating in a FastPrep-24 apparatus (MP Biomedicals) at 6 m/sec for 40 sec. Cell debris was removed by centrifugation (16,000g for 10 min at 4°C) and the supernatant was recovered. Equal OD260 nm units of each sample were layered on top of 5%–50% sucrose gradient prepared in buffer G (20 mM Tris pH 8, 100 mM NH4Cl, 15 mM MgCl2) and separated by ultracentrifugation in a Beckman SW-41Ti rotor at 39,000 rpm for 2 h 46 min at 4°C. Finally, samples were fractionated on a piston gradient fractionator (Biocomp), and the OD260 nm was recorded to generate sedimentation profiles.
Biofilm formation
Strains were cultured in BHI at 37°C overnight, diluted at OD600 nm = 0.01 in fresh medium. A 96-well plate was seeded with 100 μL and incubated at 37°C in a humidity chamber for 24 h. The mature biofilm was evaluated using crystal violet assay staining biomass. Then, 150 μL of crystal violet 1% was added for 15 min and washed using steam technology (Tasse et al. 2018). The dye was solubilized for 15 min in 200 μL of 33% acetic acid, and the OD590 nm was read in Tecan plate reader.
Eukaryotic cell and culture conditions
Human monocytes (U937) expressing the C5a receptor were previously constructed (Spaan et al. 2013). This receptor is targeted by the PVL and the γ toxin CB (HlgCB), two pore-forming toxins of S. aureus. Thus, S. aureus supernatant cytotoxicity on these cells is mainly driven by the activity of these two toxins. U937 cells were cultured in DMEM growth medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2.
Cytotoxicity assay
Bacterial strains were cultured in CCY broth at 37°C overnight, centrifuged at 10,000g for 10 min, and supernatants were collected. U937 cells were diluted at 1 × 106 cell/mL and PI was added at a final concentration of 25 μg/mL. A 96-well plate was seeded with 90 μL of this solution and 10 μL of culture supernatant was added. PI incorporation into cells was measured by determining OD at 635 nm after excitation at 535 nm using a Tecan plate reader. The measure was done every 10 min for 24 h, and the OD max was conserved to compare cytotoxic activities of the different bacterial supernatants.
Genomic analysis
All 301 clinical strains were previously sequenced, and all stages of quality control, sequence assembly, and typing, as well as genes and noncoding RNA genes analysis, were carried out (Baude et al. 2019; Gillet et al. 2021; Bastien et al. 2023). The GyrB, CoA, and RlmQ sequences corresponding to SAUSA300_0005, SAUSA300_0224, and SAUSA300_0981 of the USA300_FPR3757 reference strain were compared with the 301 clinical strains. For each gene, a global alignment of the 301 sequences with the USA300_FPR3757 reference sequence was processed with clustal omega v1.2.4 (Sievers et al. 2011). A variation parameter λ = m/n, where m represents the number of mutated amino acids and n is the number of codons in the gene (Wagner 2007), was calculated. Promotech v1.0 software (Chevez-Guardado and Peña-Castillo 2021) was used in all 301 strains for promoter detection. The DNA motif of the promoter was generated using MEME v4.11.2 (Bailey et al. 2009). Verification of the quality of the rlmQ− mutant, in particular the absence of mutations outside the rlmQ locus, has been carried out. JE2 rlmQ− transposon mutant genomic control was carried out using the isogenic WT JE2 strain. Validation of gene gain or loss was performed via a pangenome analysis and verified following the use of multiBamSummary from deepTools v3.5.0 to locate loci in the WT strain that were not covered by reads from the mutant strain. An additional analysis focusing on the presence of single nucleotide polymorphism (SNP) via variant calling was performed as in the SNP analysis section (Bastien et al. 2023).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Philippe Hammann and Johana Chicher for their help in proteomic analysis, and Isabelle Caldelari and David Lalaouna for helpful discussions. We thank Sara Moussadeq and Sylvère Bastien for their help with the bioinformatics analysis. This work was supported by the French National Research Agency ANR (SaRNAmod: ANR-21-CE12-0030-01 to S.M.; RHU IdBioriv: ANR-18-RHUS-0013 to F.V.), epiRNA grant (Region Grand Est, P.R.), and by the Interdisciplinary Thematic Institute IMCBio+, as part of the ITI 2021-2028 program of the University of Strasbourg, CNRS and Inserm, IdEx Unistra (ANR-10-IDEX-0002), SFRI-STRAT'US (ANR 20-SFRI-0012), and EUR IMCBio (ANR-17-EURE-0023) under the framework of the French Investments of the France 2030 Program. B.P.K. acknowledges support from ANR, the epiRNA funding from the Region Grand Est, the FRISBI, Instruct-ERIC, and iNEXT-Discovery infrastructures. R.B.-C. is supported by a fellowship from EUR IMCBio (ANR-17-EURE-0023).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079850.123.
-
Freely available online through the RNA Open Access option.
- Received September 27, 2023.
- Accepted November 29, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Roberto Bahena-Ceron is the first author of this paper, “RlmQ: a newly discovered rRNA modification enzyme bridging RNA modification and virulence traits in Staphylococcus aureus.” Roberto carried out this work as a PhD student at the Université de Strasbourg, working on RNA modifications in a collaborative thesis directed by Stefano Marzi and Bruno Klaholz at the Institut de Biologie Moléculaire et Cellulaire. The research topic of his thesis focused on RNA modification dynamics in the human pathogen S. aureus.
What are the major results described in your paper and how do they impact this branch of the field?
In this paper, we identified RlmQ as the enzyme responsible for the modification of 7-methyl guanosine (m7G) at position 2574 in Helix 90 of the 23S ribosomal RNA in Staphylococcus aureus (S. aureus). Interestingly, RlmQ has a similar structure to RlmI, the enzyme responsible for 5-methyl cytosine (m5C) at position 1962 in Escherichia coli (E. coli). However, it is important to note that m5C1962 is not present in S. aureus, and conversely, m7G2574 is not found in E. coli. This difference highlights how these modifications are specific to each organism. Both modifications, m7G2574 and m5C1962, are located in the tRNA accommodation corridor for the A-site. This suggests a potential connection between these methyl modifications and the correct placement of tRNA. In the functional analysis, we found that S. aureus strains where the RlmQ gene is disrupted, showed impaired virulence and increased biofilm production.
What led you to study RNA or this aspect of RNA science?
For my PhD, I searched for groups working on the structure and dynamics of RNA. This led me to focus on RNA modifications in ribosomal RNA in Staphylococcus aureus. While working on mapping rRNA modifications, I discovered that for some modified nucleotides, the RNA modification enzyme responsible was not described.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
While mapping the rRNA modifications in Staphylococcus aureus, I observed a gene that was annotated as RlmI based on sequence similarity; however, the modification produced by this enzyme was absent in S. aureus. Later, through the analysis of the presence of m7G2574 in strain mutants, I discovered that the gene, originally misannotated as RlmI, was responsible for this modification. Looking at the residues sequence close to the catalytic site we found differences between RlmI and RlmQ that are proposed to be involved in the modification specificity. Following the nomenclature for rRNA modification enzymes, we named this enzyme RlmQ.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
As a bachelor student at Universidad Veracruzana, the lectures given by Yolanda Cocotle and Rebeca Garcia on different topics of biochemistry and molecular biology showed me how amazing biochemistry can be.
If you were able to give one piece of advice to your younger self, what would that be?
One experiment at a time. Sometimes, as PhD students, we have not finished the experiment we are running when we are already thinking about the next one. If we don't take the time to deeply analyze the result, we can miss substantial information in our system.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
For my master's studies, I worked with Alfredo Torres Larios. During the time we worked together, I got inspired by his ambitious and insightful perspectives on science. During the PhD, the influence of my supervisors and the head of the unit (Pascale Romby) helped me to mature the perspective I have about science.
What are your subsequent near- or long-term career plans?
Now I'm about to finish my PhD and start a short postdoc term that will allow me to finish other projects that I worked on during my thesis. Later, I would like to try a postdoc position in a different country and then decide toward what field of science I want to move.








