
Motifs in SARS-CoV-2 evolution
- Christopher Barrett1,2,
- Andrei C. Bura1,
- Qijun He1,
- Fenix W. Huang1,
- Thomas J.X. Li1 and
- Christian M. Reidys1,3
- 1Biocomplexity Institute and Initiative, University of Virginia, Charlottesville, Virginia 22904, USA
- 2Department of Computer Science, University of Virginia, Charlottesville, Virginia 22904, USA
- 3Department of Mathematics, University of Virginia, Charlottesville, Virginia 22904, USA
- Corresponding author: duckcr{at}gmail.com
-
Handling editor: Peter Stadler
Abstract
We present a novel framework enhancing the prediction of whether novel lineage poses the threat of eventually dominating the viral population. The framework is based purely on genomic sequence data, without requiring prior established biological analysis. Its building blocks are sets of coevolving sites in the alignment (motifs), identified via coevolutionary signals. The collection of such motifs forms a relational structure over the polymorphic sites. Motifs are constructed using distances quantifying the coevolutionary coupling of pairs and manifest as coevolving clusters of sites. We present an approach to genomic surveillance based on this notion of relational structure. Our system will issue an alert regarding a lineage, based on its contribution to drastic changes in the relational structure. We then conduct a comprehensive retrospective analysis of the COVID-19 pandemic based on SARS-CoV-2 genomic sequence data in GISAID from October 2020 to September 2022, across 21 lineages and 27 countries with weekly resolution. We investigate the performance of this surveillance system in terms of its accuracy, timeliness, and robustness. Lastly, we study how well each lineage is classified by such a system.
Keywords
INTRODUCTION
Current viral genomic sequencing efforts facilitate epidemiological surveillance close to real time (Centers for Disease Control and Prevention 2021). The challenge now becomes the efficient identification of variants within these viral sequences that can pose possible threats. We present here a bottom-up approach that can rapidly predict whether or not, at a given geographical location, a lineage will dominate the viral population. This assessment only takes as input a time series of multiple sequence alignments (MSAs) consisting of genomes sampled from the viral population up to the considered point.
The key idea is the identification of site motifs: maximal sets of polymorphic sites exhibiting coevolutionary signals within the MSA, rather than considering the emergence of any one particular set of mutations. The sites that will manifest as motifs are selected on the basis of exhibiting sufficient mutational activity and satisfying a certain diversity criterion. Site motifs are then constructed using distances that can capture coevolutionary coupling between site pairs. The collection of site motifs then represents the relational structure among the considered polymorphic sites.
Finally, we introduce a scoring function that evaluates the relevancy of a lineage, with respect to the relational structure present in the alignment. Using this score, we develop a genomic surveillance system that can predict whether a lineage will dominate the viral population at a given location and a given time. Namely, an alert will be issued with respect to a lineage at a given time-location pair if we observe significant changes of the score.
We then put this framework to the test by analyzing retrospectively the sequence data obtained during the COVID-19 pandemic. Specifically, we consider SARS-CoV-2 genomics sequence data from October 2020 to September 2022, across 21 lineages and 27 countries with a weekly resolution. For the purpose of this analysis, we shall derive notions of accuracy, timeliness, and robustness and quantify its sensitivity and specificity in terms of classifying lineages at country resolutions as well as worldwide.
Background
Genomic surveillance plays an instrumental role in combating rapidly mutating RNA viruses (Gire et al. 2014). In particular, it was a vital necessity in the effective mitigation and containment of the COVID-19 pandemic (Deng et al. 2020; Robishaw et al. 2021). While mRNA vaccine development and distribution were successful in the USA, the Omicron variant still gave rise to questions regarding their efficacy (Choudhary et al. 2021). Timely vaccine development necessitates the rapid recognition of critical adaptations within SARS-CoV-2 (Poland et al. 2020; Kashte et al. 2021). Genomic surveillance leverages applications of next-generation sequencing and phylogenetic methods to detect variants that are phenotypically or antigenetically different, facilitating early anticipation and effective mitigation of potential viral outbreaks (Chen et al. 2022).
One of the central tasks in genomic surveillance is the identification of virulent emerging variants, or of variants that have developed vaccine resistance. The designation of SARS-CoV-2 variants of concern/relevance exemplifies such an identification process (Konings et al. 2021; World Health Organization 2021). This designation is based on phylogenetic methods and involves four steps: lineage assignment, mutation extraction, biological analysis, and declaration. First, a large phylogenetic tree is constructed from publicly available genomes and its subtrees are examined and cross-referenced against epidemiological information to designate new lineages (Rambaut et al. 2020; Bedford et al. 2021). Second, a collection of mutations frequently observed in a lineage is extracted and defined to be characteristic for that lineage. Third, the biological impact of this collection of mutations is then analyzed in wet-lab/in silico experiments. Last, in the wake of identified biological features, such as an increase in transmissibility or severity, the lineage/variant is declared of concern/relevance.
Population-based approaches were developed to complement phylogenetic-based methods with the goal of rapidly identifying and monitoring critical mutations on the SARS-CoV-2 genome. Frequency analysis is widely used to monitor variant circulation (Korber et al. 2020; Pachetti et al. 2020). The increasing relative frequency of a mutation might indicate the emergence of a new variant (Yin 2020). Entropy measurements, derived from nucleotide frequency, highlight nucleotide positions with high variation and facilitate the compact representation of SARS-CoV-2 variants (Fan et al. 2021).
Mutations on viral genomes do not always appear independently. For example, the S:D614G change, caused by the nucleotide mutation A23404G on the SARS-CoV-2 genome, was almost always accompanied by three other nucleotide mutations: C241T, C3037T, and C14408T (Plante et al. 2021). These four positions exhibited a coevolutionary pattern (Barrett et al. 2020; Yin 2020). In fact, it is by construction that positions in a molecule that share a common constraint do not evolve independently and therefore leave a signature in patterns of homologous sequences (Dutheil 2012; Priya and Shanker 2021). Extracting such coevolutionary signals from a sequence alignment leads to a deeper understanding of the impact of mutations on viral fitness and can facilitate the early detection of emerging variants.
Currently, coevolution detection strategies are based on observing the frequency of nucleotide combinations in two distinguished positions (Mercatelli and Giorgi 2020; Wang et al. 2021b). In protein folding, MSAs are used to identify related positions via mutual information (Tillier and Lui 2003). While there are contributions on the level of networks—for instance, Aguilar et al. (2012) studied the mutual information networks of enzymatic families in protein structures to unveil functional features—the work was focused on how to account for the effect of phylogeny on this identification (Buslje et al. 2009). To this end, two modifications to mutual information are introduced: row–column weighting (Gouveia-Oliveira and Pedersen 2007) and average product correction (Dunn et al. 2008). Mutual information has also been used to detect coevolutionary signals in alignments of RNA sequences (Gutell et al. 1992). In Gulko and Haussler (1996) and Yeang et al. (2007), statistical methods could differentiate correlation patterns induced by functional constraints from those induced by shared ancestry. These methods are concerned with pairwise relations since their objective is to determine RNA secondary structure. Thus, the idea of considering pairwise relations between columns within an MSA has been successfully used in protein and RNA folding more than a decade ago. Later, we shall see that our notions of site motifs and relational structures represent an extension of this methodology, encapsulating k-ary relations present in the MSA.
Technical overview
Co-occurring mutations exhibit traces of evolutionary selection pressure, and fitness gained through synchronized mutations can be more significant than the total fitness when each mutation occurs independently (Kauffman and Johnsen 1991; Imhof and Schlötterer 2001; Eyre-Walker and Keightley 2007). Therefore, a collection of synchronized polymorphic sites is a result of selection pressure and represents a signal for selective advantage, worth incorporating in viral surveillance frameworks.
Let A be a viral population consisting of sequences with n genomic sites (labeled as [n]: = {1, …, n}). A relational structure on the population A, denoted by 
 , is a collection of site motifs, or motifs for short, where each motif Mi ⊂ [n] consists of polymorphic sites that exhibit a similar evolutionary pattern in A, see Figure 1. Given an MSA
, is a collection of site motifs, or motifs for short, where each motif Mi ⊂ [n] consists of polymorphic sites that exhibit a similar evolutionary pattern in A, see Figure 1. Given an MSA  that consists of sampled sequences from A, we can approximate
that consists of sampled sequences from A, we can approximate  with
with  . We detail the derivation of a relational structure from a given MSA in the Materials and Methods section.
. We detail the derivation of a relational structure from a given MSA in the Materials and Methods section.

Coevolutionary pattern in an MSA (A) and the induced relational structure (B).
The relational structure of a viral population is by construction not fixed and evolves over time, due to changes in selection pressure. Drastic changes in the relational structure, as we shall see, will be indicative of watershed events in viral populations such as the emergence of a new and fitter lineage. In the following, we shall illustrate the concept, via a case study, demonstrating the evolution of relational structures in the context of viral strain competition.
Case study: evolution of the relational structure SARS-CoV-2 population in the UK
We consider the strain competition in the UK SARS-CoV-2 viral population, from October week 3, 2020 to October week 4, 2021. During this time frame, the Alpha variant (lineage B.1.1.7) emerged and quickly became the dominant strain in terms of relative frequency. After a period of time, the Alpha variant was then superseded by the Delta variant (lineage B.1.617.2), together with its AY sublineages (B.1.617.2 + AY.*), see Figure 2.

Evolution of the relational structure of the SARS-CoV-2 genome in the UK. (A) The relative frequency of key lineages (Alpha and Delta + AY.*) from October week 3, 2020 to October week 4, 2021 in the UK. (B) The relational structure of SARS-CoV-2 at: (1) November week 3, 2020, when the relative frequency of Alpha is <5%; (2) May week 1, 2021, when Delta + AY.* emerges and competes with Alpha; (3) September week 3, 2021, when the relative frequency of Delta + AY.* is >95%. This analysis only considers relational structures with a minimum motif size of five and we only display the three largest motifs.
In Figure 2, we display the relational structures at three distinct moments in time: (1) November week 3, 2020 (emergence of Alpha), (2) May week 1, 2021 (emergence of B.1.617.2 [Delta] + AY.*), and (3) September week 3, 2021 (domination of B.1.617.2 [Delta] + AY.*). In November week 3, 2020, the largest motif in the relational structure consisted of 21 sites. In fact, all of these 21 sites corresponded to characteristic mutations of Alpha. At this time, the relative frequency of Alpha is below 5%. The majority of the sequences in the populations carry the same nucleotide type as the original reference sequence on these 21 sites. However, even at low relative frequency, the characteristic mutations on these 21 sites appear in a highly coordinated fashion. As a result, these sites exhibit a similar evolutionary pattern and form a motif, see Figure 2.
In May week 1, 2021, the largest motif in the relational structure consisted of 38 sites. Among these 38 sites, 21 of them are precisely the 21 sites mentioned previously, that correspond to characteristic mutations of Alpha, while 16 of the remaining 17 sites correspond to characteristic mutations of the Delta + AY.* variants. At this time, the relative frequency of Alpha is above 90% while that of Delta + AY.* is below 5%. Note that on the 21 Alpha characteristic mutation sites, sequences in the Delta + AY.* variants are carrying the same nucleotides as the original reference sequence. On the other hand, sequences in the Alpha variants do not carry the 16 Delta + AY.* characteristic mutations. As such, the strain competition between Alpha and Delta + AY.* causes the formation of this large motif, see Figure 2.
In September week 3, 2021, Delta + AY.* becomes the dominant strain in the UK. The relative frequency of Delta + AY.* is so high that we observe little diversity in the columns of the MSA and no motifs in the relational structure.
The purpose of this paper is to provide a meaningful augmentation to existing genomic surveillance methodology. The novelty of the proposed approach lies in focusing on the linkage and not the specific patterns of mutations appearing in the data. This leads to the concept of motifs and relational structures. The proposed framework is a stepping stone to a more systematic development of the notion of relational structures for generally aligned data.
RESULTS
In this section, we present an approach based on relational structures in order to enhance genomic surveillance. Our system will issue an alert regarding a lineage, based on its effect on the relational structure. We will conduct a comprehensive retrospective analysis and investigate the performance of this surveillance system studying its accuracy, timeliness, and robustness. Finally, we will investigate how well each lineage is classified by our system.
Genomic surveillance based on relational structures
We introduce a novel measurement  (see Materials and Methods) that tracks the relevancy of a lineage L with respect to a relational structure S. The time evolution of
(see Materials and Methods) that tracks the relevancy of a lineage L with respect to a relational structure S. The time evolution of  can provide insights into the dynamics of a lineage L within the viral population. We develop a surveillance system based on the difference function
can provide insights into the dynamics of a lineage L within the viral population. We develop a surveillance system based on the difference function 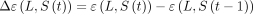 . We then perform a comprehensive retrospective analysis of the COVID-19 pandemic as well as of the performance of the surveillance
system in predicting lineage development.
. We then perform a comprehensive retrospective analysis of the COVID-19 pandemic as well as of the performance of the surveillance
system in predicting lineage development.
We investigate the development of 21 SARS-CoV-2 lineages in 27 different countries with a weekly resolution, from October week 3, 2020 to September week 4, 2022 (94 wk in total). The lineages we consider are selected among those monitored by the BV-BRC SARS-CoV-2 tracking system (Bacterial and Viral Bioinformatics Resource Center 2022; Olson et al. 2023), see Table 1. Since shared mutations among lineages transcend into similarities of their corresponding motifs, lineages are selected such that there is no substantial overlap of their characteristic mutations, see the Materials and Methods section for details of this selection procedure.
Selected lineages
The countries examined were selected by continent: We picked the top five countries from each continent with respect to their number of contributed sequences to the GISAID (Shu and McCauley 2017) database, while for Oceania we only tracked the top two; see Table 2.
Alert summary for each country, integrated over all lineages
We denote a lineage as a key lineage for a given country if its relative frequency within the viral population of that country reaches 50% at some point within our 94-wk time window. Note that the notion of key lineage is by construction country dependent.
We now describe our surveillance system based on relational structures. Its aim is to predict whether a lineage L will develop into a key lineage for the viral population X of a given country. With respect to X, an alert will be issued by our system, regarding lineage L at time t, if  , where SX(t) is the relational structure corresponding to X at time t.
, where SX(t) is the relational structure corresponding to X at time t.
Note that a lineage can trigger, for the same country, multiple alerts over a period of time. A lineage can also trigger multiple alerts across multiple countries over the same period of time.
An alert with respect to a lineage L, in a given country, will be labeled as relevant if the relative frequency of L reaches 50% within the next 3 mo in said country. Otherwise, the alert will be labeled irrelevant.
Accuracy
Integrated overall 27 countries and 21 lineages, our surveillance system issued 238 alerts in total. Of those, 136 were labeled as relevant (i.e., 57.1% accuracy). The accuracy of the alerts for all lineage-country combinations is displayed in Figure 3.

Accuracy of alerts across different variants and geographic locations. The x-axis denotes the variants considered, while the y-axis denotes the countries. The color of each dot represents the accuracy of our alert system with respect to the particular variant at the corresponding location. An empty cell indicates no alert is issued for that country lineage pair.
For the vast majority of the lineage-country combinations (437 out of 567 possible pairs), our system never issues an alert during the selected time span. Lineages B.1.1.7 (Alpha), B.1.617.2 + AY.* (Delta), and B.1.1.529 + BA.* (Omicron) are well-known for circulating globally (Tegally et al. 2020; Galloway et al. 2021; Wang et al. 2021a; Gowrisankar et al. 2022). These three lineages received alerts from our system in almost all countries and the majority of these alerts were accurate (with the exception of some alerts for Alpha in certain countries). For lineages that circulate locally, our system still accurately predicts their dominance in certain specific countries, for instance, B.1.1.519 in Mexico, B.1.351 in South Africa, and B.1.525 in Nigeria, while it remains silent in most other countries.
We proceed by investigating alerts issued for each specific country, integrated over all lineages, see Table 2.
The number of alerts varies across countries, with a mean of 8.8 and a standard deviation of 5.2. Germany has only two alerts in total, while South Africa has 25. The alert accuracy also varies: All countries in Europe exhibit an accuracy of 100.0% while those in South Africa have only 28.0% accuracy. It is worth mentioning that geographically adjacent countries tend to have similar alert patterns, which could be due to their viral populations having similar dynamics.
Finally, we investigate the alerts issued with respect to specific lineages, integrated across all countries, see Table 3.
Alert summary for each lineage, integrated over all countries
The number of alerts varies significantly by lineages, with a mean of 11.3 and a standard deviation of 19.1, with 68.1% of alerts being issued to B.1.1.7 (Alpha), B.1.617.2 + AY.* (Delta), and B.1.1.529 + BA.* (Omicron) (which were all circulating globally at their respective times). Alerts issued to B.1.617.2 + AY.* have the highest accuracy (88.7%). Finally, while alerts issued with respect to some lineages have 0% accuracy, they are sporadic and are generally issued with the “irrelevant” designation.
Timeliness
For an alert system to be useful, one would expect that relevant alerts are issued at times when their corresponding lineages exhibit low relative frequency. As such, we investigate the relative frequency of lineages that issue relevant alerts.
If there exist multiple relevant alerts for the same lineage in a given country, we consider only the first issued relevant alert for that lineage. Integrating over all countries, there are a total of 69 relevant “first” alerts. For each such alert, we investigate the relative frequency of the associated lineage in the week prior to the issuance of the alert. If the relative frequency is below 5%, we consider the alert to be timely. Of the 69 relevant “first” alerts, 42 (60.9%) are timely. The ratio of timely alerts versus “first alerts” for each country is displayed in Figure 4, while the same for each lineage is shown in Figure 5.

Timeliness of alerts across the globe. We illustrate the portion of timely first alerts (blue) versus nontimely first alerts (red).

Timeliness of alerts across lineages. Lineages that have been issued at least one relevant alert over our study period (gray box). We illustrate the portion of timely first alerts (blue) versus nontimely first alerts (red) for each of these lineages. The associated percentage is the ratio of timely first alerts to the total number of first alerts. The lineages are organized in a hierarchical tree structure according to their phylogenetic relations. Note that the 21 lineages considered in the study are somewhat independent in terms of overlaps of characteristic mutations.
Alert robustness
In the previous sections, we described various notions of the effectiveness of the enhancement of viral surveillance based on relational structures. The alerts were derived from time-stamped MSAs, where each MSA consisted of sequences collected within a specific time period for a given country. However, sequencing capabilities across different countries are vastly different: In GISAID, the average number of sequences per week in Botswana during the investigative period is 48, while the average number of sequences per week in the UK is 22,482. In view of this and in order to facilitate cross-country comparisons we study a notion of robustness of alerts.
To this end, we consider the robustness of the alerts for three lineages at three different time points in the UK. We construct
MSAs of different sizes via uniform subsampling with weekly sampling sizes of M = 50, 100, 500, and 1000. For a given time point and fixed sampling size, we construct 100 pairs of MSAs, via sampling uniformly
randomly from all the UK sequences collected within that time frame. We then investigate the consistency in alert issuance
behavior across the 100 pairs. For each sample size, the issuance of an alert can be considered as an indicator random variable
X (X = 1 for issuance and X = 0, otherwise). Then, the variance of this random variable,  , measures the inconsistency of the system's output at that sample size, see Figure 6.
, measures the inconsistency of the system's output at that sample size, see Figure 6.

Alerts and robustness with respect to different sample sizes at three different times (t1: November week 4, 2020 to December week 1, 2020, t2: November week 4, 2021 to December week 1, 2021, t3: January week 2, 2022 to January week 3, 2022). In each figure, on the z-axis we display the variance of the random variable X (X = 1 for issuance and X = 0, otherwise) associated to a lineage-sample size pair.
Note that originally, our system issued alerts for B.1.1.7 at t1, and for B.1.617.2 + AY.* and B.1.1.529 + BA.* at t2 (this was initially achieved by sampling a single alignment of size 3000). The system did not issue alerts for the remaining
time-lineage pairs. For the six combinations that correspond to this nonissuance, we observe that the system produces consistent
output ( ), even at smaller sample sizes. In fact, the system almost faithfully reproduces the original nonissuance behavior. For the
three combinations that correspond to alert issuance, we observe an increase in consistency as the sample size increases as
measured by the variance. However, the rate of increase in consistency for the three combinations varies. For B.1.617.2 +
AY.* and B.1.1.529 + BA.*, at t2 and sample size 500, the system reproduces the original issuance behavior more than 90% of the time, while the inconsistency
for the alert issuance behavior associated to B.1.1.7 at t1 is still very high (
), even at smaller sample sizes. In fact, the system almost faithfully reproduces the original nonissuance behavior. For the
three combinations that correspond to alert issuance, we observe an increase in consistency as the sample size increases as
measured by the variance. However, the rate of increase in consistency for the three combinations varies. For B.1.617.2 +
AY.* and B.1.1.529 + BA.*, at t2 and sample size 500, the system reproduces the original issuance behavior more than 90% of the time, while the inconsistency
for the alert issuance behavior associated to B.1.1.7 at t1 is still very high ( ), even at sample size 1000.
), even at sample size 1000.
To state the obvious, it is critical to include all available sequences for surveillance purposes. The emergence of new variants is difficult to observe via small array-sizes. This being said, typically 1000 sequences are sufficient to provide a robust alert. In the case of this study, the majority of countries attain this weekly sequencing rate. However, when a lineage is at very low relative frequency and the strain composition is very heterogeneous then limited sampling is simply insufficient to provide a robust alert.
Sensitivity and specificity
In the following, we shall shift focus from alerts to lineages and investigate whether or not they are correctly classified. Alerts as discussed previously are either relevant or not, while a lineage will be classified as true/false positive or true/false negative.
For a given country, we are interested in how many key lineages are successfully flagged and how many non-key lineages never
trigger an alert. The first such number measures the sensitivity of the system while the second one measures the specificity.
To this end, for a given country we denote by P the number of key lineages and by N the number of non-key lineages. The number of key lineages that trigger relevant alerts in our system will be denoted by
TP, while the number of key lineages that do not trigger relevant alerts will be denoted by FN. On the other hand, the number
of non-key lineages that never issue an alert will be denoted by TN, while the number of non-key lineages that issue any alert
will be denoted by FP. Sensitivity will then be given by TPR = TP/P, while specificity will be given by TNR = TN/N. Integrated over all countries, we have P = 81, with TP = 69 and 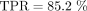 . Furthermore, we have N = 486, with TN = 429 and
. Furthermore, we have N = 486, with TN = 429 and  .
.
Sensitivity and specificity for each individual country are displayed in Table 4.
Country-specific sensitivity and specificity for the alert system
We note that TPR (sensitivity) and TNR (specificity) depend on the alert threshold θ of  . In general, if θ decreases, additional alerts will be issued and hence it is more likely that key lineages will be flagged
by relevant alerts. This will lead to an increase in TP and hence an increase in sensitivity. However, at the same time, TN
will decrease as it is more likely that non-key lineages will be flagged by alerts, which in turn reduces specificity. To
quantify this trade-off, we perform a receiver operating characteristic (ROC) analysis (Fawcett 2006). Each θ induces a pair (FPR, TPR), where FPR = 1 − TPR. Varying θ ∈ [0, 1] produces the ROC curve (Fawcett 2006), where the x-axis and y-axis represent the FPR and TPR, respectively. ROC curves for each of the countries considered, as well as a global ROC curve
(integrated over all countries considered), are displayed in Figure 7.
. In general, if θ decreases, additional alerts will be issued and hence it is more likely that key lineages will be flagged
by relevant alerts. This will lead to an increase in TP and hence an increase in sensitivity. However, at the same time, TN
will decrease as it is more likely that non-key lineages will be flagged by alerts, which in turn reduces specificity. To
quantify this trade-off, we perform a receiver operating characteristic (ROC) analysis (Fawcett 2006). Each θ induces a pair (FPR, TPR), where FPR = 1 − TPR. Varying θ ∈ [0, 1] produces the ROC curve (Fawcett 2006), where the x-axis and y-axis represent the FPR and TPR, respectively. ROC curves for each of the countries considered, as well as a global ROC curve
(integrated over all countries considered), are displayed in Figure 7.

ROC curves across the globe. The x-axes and y-axes represent FPRs and TPRs, respectively.
DISCUSSION
In this paper, we present an augmentation of genomic surveillance via a novel approach. The concept of motifs and relational structure is shown to provide at times accurate and timely predictions with respect to the development of viral lineages.
The key idea behind this approach is the study of site co-occurring mutational patterns that quantify MSA evolution. This is not new; however, what is novel is the focus not on the specific pattern but on the relational linkage between sites. Genomic adaptation is, to a large extent, not facilitated by isolated mutations but rather by collections of mutations. Functionally connected mutations changing simultaneously therefore provide a key indicator of relevant evolutionary dynamics, and even at low frequencies, co-occurring mutations are indicative of the existence of noteworthy biological mechanisms inducing differentials in said dynamics.
The relational structure notion introduced here captures maximal sets of sites that simultaneously experience selection pressure. Differential changes of the relational structure within the MSA provide crucial information about the viral “heartbeat” since these are induced by selection: typically changes for the worse from the perspective of the viral host.
In the following, we will demonstrate how the relational structure approach can complement existing methods. We will first explore its impact from a data reduction standpoint and then from a perspective that considers the duality between sequences and sites.
Data reduction
In the realm of SARS-CoV-2 genomic surveillance, progress in sequencing technology coupled with global collaboration among scientists has enabled us to acquire comprehensive viral genome sequences at an unprecedented speed. For instance, during its peak, the United States was amassing more than 100,000 sequences every week (Mercatelli and Giorgi 2020). Due to the rapid mutability of viral sequences and the extensive sample size, the collected sequence data often displays numerous distinct mutations. Many of these mutations are inherently noisy, as a substantial portion of them arises from random, isolated occurrences. An essential challenge in contemporary genomic surveillance is to identify critical mutations that correspond to the emergence of more advantageous lineages. This is particularly relevant when these mutations are still at relatively low frequency, necessitating timely alerts for informed responses.
Due to the combinatorial explosion, monitoring and testing for each possible combination of mutations within the evolving viral population quickly become impractical. Thus, data reduction methods are needed to narrow down the search space. Currently, leveraging expert domain-specific knowledge constitutes one of the principal (empirical) methods of data reduction and synthesis. In current surveillance systems, data reduction leveraging biological insights tends to focus on regions that have already been established as biologically significant. For instance, the National SARS-CoV-2 Strain Surveillance (NS3) program (Lambrou et al. 2022) incorporates mutations on epitopes in the spike protein as part of its routine. Nevertheless, mutations occurring beyond these areas could also be of crucial importance. To illustrate this point, consider the subvariants BA.4 and BA.5 of the SARS-CoV-2 Omicron variant. These two lineages differ exclusively outside the spike region (Tegally et al. 2022) and hence are not distinguishable by an epitope-focused approach. However, BA.5 is the more transmissible of the two, causing 65% of cases, while BA.4 accounts for only 17% (World Health Organization 2022). In this specific example, an exclusively epitope-focused approach could have potentially missed the emergence of BA.5.
The relational structure approach presented in this paper can be used as an alternative, data-driven reduction method that does not rely on a priori biological knowledge, complementing existing methods. To demonstrate this point, let us revisit the BA.4/BA.5 example. The relational structures of BA.4 and BA.5, when their corresponding motif-based alerts were triggered, exhibit significant differences in mutations outside the spike region, see Figure 1 in Barrett et al. (2022). Specifically, the BA.5 alert involves a distinguishing cluster of seven sites, one of which exhibits D3N on the membrane protein, three encoding ORF6:D61L, and the remaining three exhibiting the synonymous mutations C26858T, C27889T, and A27259C (Barrett et al. 2022). Therefore, motif analysis can enhance existing approaches by unveiling the linkages between spike and non-spike protein sites, see Figure 2, serving as a rapid, automatic, and cost-efficient filtering step for downstream biological analysis.
Sequence-site duality
The framework of relational structures can be seen as a distinct but complementary approach to existing sequence-centric genomic surveillance. Phylogenetic analysis and lineage assignment techniques focus on the evolutionary relatedness of genomic sequences within the MSA. Metric-based tree inference techniques like quartet mapping (Eigen et al. 1988) and split decomposition (Bandelt and Dress 1992) can be understood as recursive hierarchical clustering methods. A lineage can then be conceptualized as a subtree of a phylogenetic tree. On the other hand, the relational structure encompasses the interactions between sites in the MSA. A motif within a relational structure is thus comprised of sites that exhibit proximity based on an information-theoretic distance measure. In this paper, we used a straightforward clustering method to establish these motifs. Alternatively, more advanced metric-based tree inference methods can be used on sites while still utilizing the type of information distance between them. This approach would, when subjected to hierarchical clustering, result in the concept of a “dual tree,” that is, a tree on the MSA's sites. Interpreting such a dual tree, however, would necessitate further investigation.
As we demonstrated in the Results section, combining the relational structure with lineage information, in the form of characteristic
mutations, can provide us with deeper insights into how the lineage itself emerges and develops. Our score  captures by design the impact a lineage has on the formation of large motifs within the relational structure, and thus can
provide information beyond characteristic mutation frequencies by capturing the degree of coordination between said characteristic
mutations. A high degree of coordination provides a sensible statistical signal which can facilitate timely predictions.
captures by design the impact a lineage has on the formation of large motifs within the relational structure, and thus can
provide information beyond characteristic mutation frequencies by capturing the degree of coordination between said characteristic
mutations. A high degree of coordination provides a sensible statistical signal which can facilitate timely predictions.
One of the key challenges in combining the relational structure approach with lineage information is that they are in principle not always compatible. In our approach, they are linked via the characteristic mutations corresponding to each lineage. For each lineage, we obtained the characteristic mutation list from the Pango database (Rambaut et al. 2020). However, currently there are no generally agreed upon universal rules to define the characteristic mutations of a lineage. For a given lineage, the list of characteristic mutations can differ across different databases and each database has its own rules for defining characteristic mutations. Furthermore, characteristic mutations are not “characteristic” enough with the same mutation sometimes being used to characterize multiple lineages (Hodcroft 2021). This is particularly the case when one lineage is a sublineage of the other. To reduce the impact introduced by the hierarchical structure within the lineages themselves, for this paper we selected and merged lineages so that the sequences became better partitioned. In subsequent publications, we will address the natural question that arises from this consideration, namely how to address hierarchical structures within designated lineages.
Currently, there is no universal system that performs “well” for viral taxonomy below the level of species. In Rambaut et al. (2021), a dynamic nomenclature approach was proposed for SARS-CoV-2 lineages based on phylogenetic methods. However, their approach still has the issue of over-designation, as more than 2500 lineage/sublineage names are assigned (Rambaut et al. 2020) and only a handful of them are considered to be critical from an epidemiological perspective. Our hope is that, by bringing relational structures into the picture, we can identify key lineages in a more accurate and timely fashion.
In fact, this approach can be generalized to a comprehensive system for classifying genetic diversity within a population and designating (sub)lineages. The key to such a general system is inferring the hierarchical (sub)lineage structure from the relational structures. More precisely, one could use each relational structure to construct a partition of the corresponding sequence population into nonoverlapping subsamples that exhibit similar evolutionary patterns which support said relational structure. For each subsample, one then recursively computes its relational structure, which in turn yields (sub)lineages for the given subpopulation. By this procedure, one could recover the hierarchical structure between lineages in an effective manner and without specialized human input, as is the case for Pango.
MATERIALS AND METHODS
Lineage selection
We begin with all 28 lineages of concern that are currently or have been previously monitored by the BV-BRC SARS-CoV-2 tracking system (Bacterial and Viral Bioinformatics Resource Center 2022; Olson et al. 2023). A lineage of concern (LOC) is a group of closely related viral sequences that exhibit similar mutations or recombinations that may significantly affect vaccine efficacy, transmissibility, or disease outcomes (Bacterial and Viral Bioinformatics Resource Center 2022). The designation of LOCs is based on phylogenetic methods, and cross-referenced against epidemiological information (Bedford et al. 2021; Rambaut et al. 2021).
According to Nextstrain and CoVariants (Hadfield et al. 2018; Hodcroft 2021), these lineages exhibit a hierarchical structure which reflects the evolutionary trajectories the viral population undertook when acted upon by selection pressures, see Figure 8. For example, BA.1 and BA.2 are sublineages of Omicron; BA.4 and BA.5 are sublineages of BA.2. Such a hierarchical structure affects the lineage assignment of viral genomic sequences, as sequences in a sublineage are also assigned to their ancestors. This lineage assignment method has been adopted by the CDC during their genomic surveillance efforts (CDC 2021). However, this is not the case for the GISAID database (Shu and McCauley 2017), where each sequence is assigned when uploaded to a single lineage based on which characteristic mutations it carries.

The hierarchical structure of the 28 lineages and sublineages monitored by the BV-BRC SARS-CoV-2 tracking system (Bacterial and Viral Bioinformatics Resource Center 2022). This structure reflects their evolutionary trajectories and relationships (Hadfield et al. 2018; Hodcroft 2021). The lineages selected for our study (green) are independent in this hierarchy structure.
Our approach takes the hierarchical structure of the 28 LOCs into account by stratifying them. In this study, we only consider one level of this hierarchy, which consists of 21 lineages, with Omicron (B.1.1.529 + BA.*) including all BA.* sublineages, see Figure 8. Our choice of these 21 lineages allows us to partition the sequence space into nonoverlapping subsets, facilitating the downstream analysis on lineage relative frequency calculations. We follow the characteristic mutation definitions given in Pango Lineages (Rambaut et al. 2020) for our considered LOCs.
Data preparation
SARS-CoV-2 whole genome data was collected from GISAID (Shu and McCauley 2017). All collected sequences were aligned to the reference sequence collected from Wuhan, 2019 (GISAID ID: EPI_ISL_402124). An MSA was produced by MAFFT (Katoh et al. 2002). Each sequence was labeled by collection time, country and classified by lineage.
We partitioned the sequences into time bins according to their collection times at a weekly resolution, where week 1 equals days 1–7, week 2 equals days 8–15 and the following weeks are mapped accordingly. We then integrate sequences over three consecutive time bins into one MSA, labeled by the last time bin. In countries like the UK and the USA, there were more than 10,000 sequences per week when the virus is heavily circulated in the human population. In view of computational feasibility, we implemented a sequence cap of 3000 per week via uniform sampling.
The relative frequency for a lineage within the population at a given time was computed based on the lineage classification in the respective time bin. If the sequence was labeled by one of the 21 selected lineages, it was counted as its respective lineage. However, if a sequence was labeled by a sublineage of one of the 21 selected lineages, then it contributed to the frequency of its parent among the 21 lineages.
Relational structure construction
For a given MSA, the relational structure is constructed by first selecting polymorphic sites, and then partitioning the polymorphic sites into motifs via clustering.
Polymorphic sites selection
Let M be an MSA with m sequences of length n, where each row is a sequence, and each column corresponds to a genetic site. We filter for sites exhibiting sufficient large
nucleotide diversity such that a significant coevolutionary signal may be observed. To this end, we utilize Shannon entropy
(Shannon 1948) to identify sites where selection induces evolutionary variation. Namely,
 where the units of H are bits, and pi(x) is the probability of the nucleotide x appearing in column i. Note that there may be gaps in the column of an MSA. If a column contains more than 30% gaps, this column will be filtered
out and not considered for further calculations. Otherwise, we ignore the gaps and compute the entropy of the columns based
on the remaining nucleotide identities. A site i is selected if H(i) > h0. Here, we set h0 = 0.1.
where the units of H are bits, and pi(x) is the probability of the nucleotide x appearing in column i. Note that there may be gaps in the column of an MSA. If a column contains more than 30% gaps, this column will be filtered
out and not considered for further calculations. Otherwise, we ignore the gaps and compute the entropy of the columns based
on the remaining nucleotide identities. A site i is selected if H(i) > h0. Here, we set h0 = 0.1.
Computing pairwise J-distance between selected sites
We then construct a distance via joint entropy and mutual information as follows: The joint entropy H(i, j) of two sites i and j is defined as
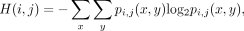 where pi,j denotes the joint distribution of columns i and j, that is, pi,j(x, y) specifies the probability of pairs of nucleotides (x, y) ∈ {A, T, C, G} × {A, T, C, G}. Here, we will not consider as a pair x, y if either x or y is a gap. Clearly, the marginal probability distributions for columns i and j are given by
where pi,j denotes the joint distribution of columns i and j, that is, pi,j(x, y) specifies the probability of pairs of nucleotides (x, y) ∈ {A, T, C, G} × {A, T, C, G}. Here, we will not consider as a pair x, y if either x or y is a gap. Clearly, the marginal probability distributions for columns i and j are given by  and
and  , respectively.
, respectively.
The mutual information I(i;j) between sites i and j is the relative entropy between the joint distribution pi,j(x, y) and the product distribution pi(x)pj(y):
 where D(p||q) denotes the Kullback–Leibler divergence of the distribution p with respect to the distribution q (Kullback and Leibler 1951). The mutual information I(i;j) quantifies the amount of information shared by two columns i and j.
where D(p||q) denotes the Kullback–Leibler divergence of the distribution p with respect to the distribution q (Kullback and Leibler 1951). The mutual information I(i;j) quantifies the amount of information shared by two columns i and j.
The J-distance represents the information-theoretic counterpart of the Jaccard distance (Jaccard 1912; Cover and Thomas 2006). J(i, j) satisfies the following properties: 0 ≤ J(i, j) ≤ 1, J(i, j) is a pseudometric, that is, J(i, i) = 0, J(i, j) = J(j, i) and the triangle inequality J(i, j) ≤ J(i, k) + J(k, j). Furthermore, J(i, j) is scale invariant, that is, J(i, j) is independent of the MSA size and uniquely determined by the joint distribution of pairs of bases.
Constructing motifs via clustering
We utilize highly connected subgraphs (HCSs) clustering (Hartuv and Shamir 2000) to compute the relational structure based on the pairwise J-distances for the selected pairs of sites. The HCS clustering algorithm is based on the partition of a similarity graph into all its HCSs. More precisely, two active columns v1, v2 ∈ V are in the same cluster if they belong to the same HCS of G. HCS clustering does not make any a priori assumptions on the number of clusters. Two sites are clustered purely based on their pairwise J-distance and it is irrelevant to how many sites are to be clustered. Therefore, the clustering result is robust with respect to the selection of columns with diversity (different choices of h0 do not impact whether the same two sites cluster together).
Based on the pairwise J-distance for the selected sites, we construct the similarity graph G = (V, E) as follows: Two vertices vi, vj ∈ V are connected by an edge if their J-distance is smaller than the threshold J(i, j) < m0. In this paper, we set m0 = 0.6. We then group all selected sites into disjoint clusters (motifs) M1, M2, …, Mr via HCS clustering on the similarity graph. Here, we only consider clusters with size ≥5, as most of the currently designated lineages contain at least five characteristic mutations. This yields the relational structure of a given MSA S = {M1, M2, …, Mr} as collection of clusters.
Relevancy assessment of a lineage
A lineage L is a collection of characteristic mutations, L = {μ1, …, μk}. We will introduce a score,  , for assessing the relevancy of L by a relational structure S = {M1, M2, …, Mr}. Let i be a site and consider a mapping Q(i), where Q(i) = |Mj| if i ∈ Mj for some 1 ≤ j ≤ r. Otherwise, Q(i) = 0.
, for assessing the relevancy of L by a relational structure S = {M1, M2, …, Mr}. Let i be a site and consider a mapping Q(i), where Q(i) = |Mj| if i ∈ Mj for some 1 ≤ j ≤ r. Otherwise, Q(i) = 0.
Let  be all sites contained in S, and Y = {y1, …, yk} where the mutation μi takes place at site yi. We then have
be all sites contained in S, and Y = {y1, …, yk} where the mutation μi takes place at site yi. We then have

Alert issuance
Our surveillance system aims to predict whether a lineage L will develop into a key lineage for a given country X. With respect to X, an alert will be issued by our system, regarding lineage L at time t, if  , where SX(t) is the relational structure corresponding to X at time t.
, where SX(t) is the relational structure corresponding to X at time t.
DATA DEPOSITION
The nucleotide sequences of the SARS-CoV-2 genomes used in this analysis are available, upon free registration, from the GISAID database (https://www.gisaid.org/).
ACKNOWLEDGMENTS
We thank Dr. Anindya Dutta and Briana Wilson for the discussion and inspiration for this work. We thank Dr. Andrew Warren for helping us access SARS-CoV-2 data. Dr. Warren pointed out the importance of applying our method to LOC, monophyletic clusters and provided us with numerous references. We thank Mia Shu for helping us to access and process the GISAID data. Many thanks to Maxwell Reidys for his proofreading. This work was partially supported by the Virginia Department of Health (VDH) grant PV-BII VDH COVID-19 Modeling Program VDH-21-501-0135.
Author contributions: C.B. and C.M.R. designed the research; F.W.H. and T.J.X.L. collected data; A.C.B., Q.H., F.W.H., T.J.X.L., and C.M.R. performed research and analysis; C.B., A.C.B., F.W.H., Q.H., T.J.X.L., and C.M.R. wrote the paper.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079557.122.
- Received December 15, 2022.
- Accepted September 20, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








