
No evidence for epitranscriptomic m5C modification of SARS-CoV-2, HIV and MLV viral RNA
- 1Institute of Molecular Biology, Biocenter, Medical University of Innsbruck, Innsbruck 6020, Austria
- 2Institute of Virology, Department of Hygiene, Microbiology and Public Health, Medical University of Innsbruck, Innsbruck 6020, Austria
- 3Institute of Bioinformatics, Biocenter, Medical University of Innsbruck, Innsbruck 6020, Austria
- Corresponding author: Alexandra.lusser{at}i-med.ac.at
Abstract
The addition of chemical groups to cellular RNA to modulate RNA fate and/or function is summarized under the term epitranscriptomic modification. More than 170 different modifications have been identified on cellular RNA, such as tRNA, rRNA and, to a lesser extent, on other RNA types. Recently, epitranscriptomic modification of viral RNA has received considerable attention as a possible additional mechanism regulating virus infection and replication. N6-methyladenosine (m6A) and C5-methylcytosine (m5C) have been most broadly studied in different RNA viruses. Various studies, however, reported varying results with regard to number and extent of the modification. Here we investigated the m5C methylome of SARS-CoV-2, and we reexamined reported m5C sites in HIV and MLV. Using a rigorous bisulfite-sequencing protocol and stringent data analysis, we found no evidence for the presence of m5C in these viruses. The data emphasize the necessity for optimizing experimental conditions and bioinformatic data analysis.
Keywords
INTRODUCTION
The SARS-CoV-2 pandemic has created an immense surge of virus-related research activity. Large efforts have been directed toward the elucidation of the structure of the SARS-CoV-2 genome as well as the RNA and protein products made from it. Nine dominant subgenomic (sg) sense RNAs are transcribed from the ∼30 kb long positive-strand genomic RNA, all of which share the same 3′UTR and have a poly(A) tail that can vary in length (Kim et al. 2020). Most but not all of the predicted open reading frames in the viral sgRNAs are translated into proteins (Jungreis et al. 2021). Furthermore, the viral RNA interacts with a broad range of viral as well as host cell proteins, and several viral RNA binding proteins (RBPs) associate with host mRNAs (Schmidt et al. 2020; Kamel et al. 2021). In addition, a number of studies have examined potential epitranscriptomic modifications of the SARS-CoV-2 genomic and sgRNAs in an effort to better understand the regulation of virus infectivity and proliferation.
In terms of variety and abundance, epitranscriptomic modifications are most widespread in tRNA and rRNA, while so far only a handful of modifications has been reported for mRNA of eukaryotic cells (Wiener and Schwartz 2021; Moshitch-Moshkovitz et al. 2022; Motorin and Helm 2022). RNA base modifications have also been detected in various RNA and DNA viruses with N6-methyladenosine (m6A) appearing as the most prevalent type (Motorin and Helm 2022). In addition, pseudouridine (ψ), adenosine to inosine (A-to-I) editing and 5-methylcytosine (m5C) among others have been reported (McIntyre et al. 2018; Courtney 2021; Ruggieri et al. 2021; Izadpanah et al. 2022). In SARS-CoV-2 viral RNA, between four and 27 m6A sites were identified by different studies (Burgess et al. 2021; Campos et al. 2021; Li et al. 2021; Liu et al. 2021; Zhang et al. 2021; Izadpanah et al. 2022). Moreover, ψ, A-to-I editing and m5C have been found (Taiaroa et al. 2020; Fleming et al. 2021; Picardi et al. 2021; Lin et al. 2022). Collectively, however, the extent of the epitranscriptome as well as the functional significance of RNA base modifications in SARS-CoV-2 remain poorly understood (Izadpanah et al. 2022).
In particular, the existence and function of m5C in SARS-CoV-2 RNA and other RNA viruses remains ambiguous. A study using Nanopore direct RNA sequencing (DRS) of human coronavirus HuCoV-229E RNA reported numerous putative m5C sites distributed along sgRNAs (Viehweger et al. 2019). Using the same approach with SARS-CoV-2, Kim et al. (2020) detected 41 potential modifications but, apart from excluding m5C, did not elucidate their nature. In contrast, another DRS study published in bioRxiv reported the identification of 42 m5C positions in sgRNAs (Taiaroa et al. 2020), and a study applying the bisulfite-sequencing technique detected 555 high confidence m5C sites in SARS-CoV-2 RNA isolated from Vero E6 cells (Lin et al. 2022). Divergent claims regarding the abundance of m5C have also been made for human immunodeficiency virus (HIV) and murine leukemia virus (MLV; Courtney et al. 2019a,b; Eckwahl et al. 2020; Cristinelli et al. 2021). Functional studies showed that abrogation of the cytosine methyltransferase NSUN2 inhibited alternative virus RNA splicing and translation of HIV proteins and reduced expression of MLV proteins (Courtney et al. 2019a,b). The latter, however, was not observed by another study, which instead found that MLV protein and virion production was sensitive to knockdown of the mRNA nuclear export factor and m5C reader protein ALYREF (Eckwahl et al. 2020).
In light of the conflicting results with respect to the number and positions of m5C in viral RNA, we set out to examine m5C in SARS-CoV-2 RNA as well as to confirm previously reported cytosine methylation in the RNA of HIV and MLV (Courtney et al. 2019a,b; Eckwahl et al. 2020; Cristinelli et al. 2021) using a rigorous bisulfite-sequencing analysis protocol. Our results argue against the presence of epitranscriptomic cytosine modification in all three viruses.
RESULTS AND DISCUSSION
Bisulfite-sequencing analysis of SARS-CoV-2 RNA
Treatment of isolated RNA with bisulfite leads to the deamination of cytosine (C) to uracil (U) at controlled pH and temperature conditions, while C that is methylated at the carbon 5 atom is largely protected from this reaction (Frommer et al. 1992). Sequencing of the treated RNA, therefore, allows for the selective detection of methylated and unmethylated Cs. One big caveat of this method, however, is the inhibition of cytosine deamination if the C is in a double strand conformation (see Trixl and Lusser 2019 for an in-depth discussion). Thus, failure to resolve secondary structures or RNA duplexes (e.g., between sense and antisense RNA) before bisulfite treatment can result in the occurrence of false positive m5C calls. Similar to other RNA viruses, SARS-CoV-2 genomic and sgRNAs form pronounced secondary and tertiary structures (Andrews et al. 2021). Moreover, virus replication and transcription of sgRNAs involves the generation of an antisense transcript that serves as template for the production of those RNAs (Kim et al. 2020). Thus, the bisulfite treatment conditions have to be carefully optimized for the analysis of viral RNA in order to prevent detection of false positives. To validate m5C sites in SARS-CoV-2 RNA previously detected by DRS, we selected a 316 nt region in the ORF of the nucleocapsid (N) protein that was reported to contain eight methylated Cs (Taiaroa et al. 2020) for analysis by bisulfite-sequencing (Fig. 1A). Total RNA was isolated 48 h after infection with SARS-CoV-2 D614G variant from African green monkey Vero E6 cells stably overexpressing human transmembrane protease serine subtype 2 (TMPRSS2) and ACE2 receptor as well as from infected human Caco-2 cells. The RNA was subjected to our optimized bisulfite treatment protocol followed by cDNA synthesis, PCR amplification and subcloning of the selected region. Sanger sequencing of multiple individual clones revealed C-to-T conversion of all cytosines in all clones independent of the cell line pointing toward the absence of m5C in this region (Fig. 1B,C). In contrast, the well-known methylation sites C38, C47, and C48 in human tRNAAsp were readily detected with our protocol (Fig. 1C).
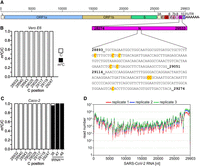
Bisulfite-sequencing analysis reveals no m5C sites in SARS-CoV-2 RNA. (A) Schematic representation of SARS-CoV-2 genome structure. A 316 nt sequence stretch of the N protein-encoding region harboring eight putative m5C sites (Taiaroa et al. 2020) is highlighted, and the m5C candidate sites are marked in red (numbers corresponding to NC_045512v2 sequence). (B,C) Bisulfite-treated RNA from virus-infected Vero E6-TMPRSS2-ACE2 (B) or human Caco-2 cells (C) was reverse transcribed, and the region indicated in (A) was amplified by PCR followed by subcloning and Sanger sequencing of individual clones (n = 35 for each cell line). No unconverted Cs were detected at the indicated positions in viral RNA. Methylated positions C38, C47, and C48 in human tRNAAsp are shown as positive control (n = 20). (D) Read coverage of SARS-CoV-2 RNA in three biological replicates after RNA-seq of bisulfite-treated RNA from infected Vero E6-TMPRSS2-ACE2 cells.
To examine the m5C pattern at a transcriptome-wide level and with higher coverage compared to the PCR protocol, we subjected isolated poly(A) RNA from infected Vero E6-TMPRSS2-ACE2 cells to bisulfite treatment, library preparation and deep sequencing. Agilent Bioanalyzer results of the poly(A)RNA revealed strong enrichment of the abundant viral sgRNAs demonstrating robust infection of the cells (Supplemental Fig. S1). To control for the efficiency of C-to-U conversion, unmethylated ERCC standard RNA was spiked into the samples before bisulfite treatment and deep sequencing. For a rigorous analysis of the sequencing data, we applied a modified version of the meRanTK software (version 1.3.0). Thereby, the meRanCall module of the original analysis workflow (Rieder et al. 2016) was updated to include additional stringency filters (see Materials and Methods) to avoid false positive m5C calling. Deep sequencing resulted in extensive read coverage across the entire virus genome as well as the ERCC standards (Fig. 1D; Supplemental Fig. S1). The 3′end in particular was covered by a large number of reads reflecting the transcription of sgRNAs from this part of the genome (Fig. 1D; Kim et al. 2020). Importantly, however, no high confidence m5C candidate sites were called using our stringent filtering parameters. These results, therefore, argue against the presence of significant methylation in genomic or subgenomic RNA of SARS-CoV-2.
Our findings deviate from an earlier bisulfite study reporting the identification of several hundred m5C sites (Lin et al. 2022). As we used the same host cells and a comparable virus variant, the most likely explanation for the observed discrepancy is the use of different bisulfite treatment and data analysis procedures. Although we used the same commercial kit for bisulfite conversion as Lin et al. we have implemented additional steps to optimize deamination and desulfonation efficiency (see Materials and Methods). In particular, we added formamide and performed three temperature cycles to improve RNA double strand melting. Moreover, improved desulfonation is accomplished by incubation at 37°C instead of room temperature. In this way, we achieved a C conversion frequency of 99.9% (calculated from ERCC), while Lin et al. reported 97.37%. The latter would account for about 260 methylation-independent, nonspecific, nonconversion events per 10 kb sequence or about 790 for the whole virus genome. Furthermore, our bioinformatic data analysis workflow included stringency filters specifically tailored to the analysis of RNA methylation data (see Materials and Methods; Rieder et al. 2016; Huang et al. 2019), while Lin et al. (2022) used low stringency parameters and software typically applied to DNA methylation analyses.
The difference between our results and previously published DRS studies is most likely owed to the still young DRS technology and the difficulties with accurate interpretation of modification-related signals (Anreiter et al. 2021; Begik et al. 2022). While two DRS studies found apparent m5C positions in their analyses of SARS-CoV-2 and HuCoV-229E (Viehweger et al. 2019; Taiaroa et al. 2020), it was later shown that unmethylated in vitro generated transcripts of specific SARS-CoV-2 genomic regions elicited similar ionic current signals as native viral transcripts, thereby ruling out the presence of m5C across the tested regions (Kim et al. 2020).
Bisulfite-sequencing analysis of HIV and MLV RNA
HIV and MLV are two other RNA viruses for which cytosine methylation was reported recently. In the case of HIV, m5C had been examined by either using an immunoprecipitation-based (meRIP) protocol (Courtney et al. 2019b) or a bisulfite-sequencing approach (Cristinelli et al. 2021). While the former study found 19 distinct methylation peaks spread along the entire virus genome, the latter analysis detected seven high confidence sites in the vicinity of the gag-pol ribosomal frameshift signal that were present at all points of a time course experiment of HIV infection. To confirm these sites, we performed bisulfite treatment of RNA extracted from human PM1 cells infected with HIV NL4-3 and spiked with ERCC standard RNA followed by PCR-mediated amplification of the region spanning the gag-pol frameshift signal. In addition, we amplified a region toward the 3′end of HIV gRNA that harbored m5C peaks in a previous study (Fig. 2A; Courtney et al. 2019b). The results reveal the expected high methylation rates at positions C38, C47, and C48 of human tRNAAsp that we used as the internal positive control, as well as the absence of any nonconversion event in ERCC-derived clones (negative control; Fig. 2B). However, we could not verify methylation of the previously reported m5C candidate sites neither in the gag-pol region nor in the region toward the 3′end of HIV RNA (Fig. 2B). These results were quite unexpected as the bisulfite sequencing and data analysis protocol that Cristinelli et al. (2021) used was very similar to our method. An important difference, however, lies in the additional filtering of the m5C candidate sites to improve the signal-to-noise ratio that we included in the updated version of the meRanTK software. Additionally, we used a replication competent HIV NL4-3 isolate, whereas Cristinelli et al. used an HIV-based vector system pseudotyped with VSV-G. Although in both cases the utilized human T cell line (PM1 versus SupT1) produced viral RNAs, only in our study full replication of the virus occurred with production of infectious viral progeny and secondary infection. However, we also examined the gag-pol region in a different HIV isolate (D117/II) and received the same negative result (Supplemental Fig. S2).
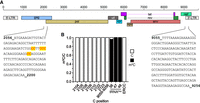
Bisulfite-sequencing analysis of previously reported m5C sites in HIV RNA. (A) Schematic representation of HIV genome structure. Amplicon sequences harboring seven previously reported m5C candidate sites (marked in red) in the gag-pol region (left; Cristinelli et al. 2021) and amplicon sequence covering previously reported meRIP peaks (right; Courtney et al. 2019b) are shown (numbers corresponding to AF324493.2 sequence). (B) Bisulfite-treated RNA from virus-infected PM1 cells was reverse transcribed, and the regions indicated in (A) were amplified by PCR followed by subcloning and Sanger sequencing of individual clones (n = 35). No unconverted Cs were detected at the indicated positions in viral RNA. For amplicon 9055–9256, no unconverted C was detected over the whole range. Methylated positions C38, C47, and C48 in human tRNAAsp are shown as positive control (n = 20); results from the spike-in ERCC136 RNA (nt 81–323) are shown as negative control (n = 10).
Cytosine methylation has also been reported for MLV. As for SARS-CoV-2 and HIV, the numbers of reported sites varied greatly. While meRIP revealed about 40 m5C peaks, only five high confidence sites were identified using bisulfite-sequencing (Courtney et al. 2019a; Eckwahl et al. 2020). We therefore designed primers for the amplification of the regions spanning the reported sites (Eckwahl et al. 2020) along with a region harboring several meRIP peaks (Fig. 3A; Courtney et al. 2019a) and examined the bisulfite-treated RNA from MLV-infected Dunni cells by Sanger sequencing of individual clones. Similar to the results obtained for SARS-CoV-2 and HIV, all scrutinized cytosines were completely converted to uracil indicating the absence of cytosine methylation in MLV RNA, while the known methylations in tRNAAsp from the same cells were readily detectable (Fig. 3B). As pointed out above, technical issues are most likely the cause for these differences. For instance, the specificity of meRIP is strongly contingent on the quality of the m5C antibody as shown recently in a comparative study (Weichmann et al. 2020). Because no details are given if and how the issue of false positive m5C calls was addressed in the study by Eckwahl et al. (2020), it is difficult to explain the observed discrepancy. We note that we have used the Friend MLV strain rather than the pNCA/1-9219 plasmid used by Eckwahl et al. which in theory could give rise to the differences between the two studies even though this option is unlikely because of the high degree of sequence conservation (four out of five m5C candidate sites). To better understand the reason for the discrepancies, we reanalyzed the original bisulfite-sequencing data sets (Eckwahl et al. 2020; Cristinelli et al. 2021) for HIV and MLV RNA with meRanTK v.1.3.0 using the same settings as for our SARS-CoV-2 analysis (note that no raw data are publicly available for the Lin et al. study). While we detected between 40 and 115 m5C sites in the host transcriptomes of MLV and HIV infected cells (present in two replicates), respectively, including the well-characterized methylation sites in mitochondrial 16S rRNA (m5C911 in mouse, m5C1488 in human, Supplemental Table S1), no m5C was detected in the reported viral RNA regions. These results support our experimental analyses in the respective viruses using PCR-bisulfite sequencing (Figs. 2, 3) and suggest that experimental and analysis procedures rather than strain particularities are responsible for the different results.
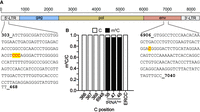
Bisulfite-sequencing analysis of previously reported m5C sites in MLV RNA. (A) Schematic representation of MLV genome structure. Amplicon sequences harboring four previously reported (Eckwahl et al. 2020) m5C candidate sites (marked in red) toward the 5′ end (left) and the 3′ end (right), respectively, are shown (numbers corresponding to NC_001362.1 sequence). (B) Bisulfite-treated RNA from virus-infected Mus dunni cells was reverse transcribed, and the regions indicated in (A) were amplified by PCR followed by subcloning and Sanger sequencing of individual clones (n = 35 each). No unconverted Cs were detected at the indicated positions in viral RNA. Methylated positions C38, C47, and C48 in mouse tRNAAsp are shown as positive control (n = 10); results from the spike-in ERCC136 RNA (nt 81–323) are shown as negative control (n = 10).
Even though our results argue against wide-spread modification of SARS-CoV-2, HIV and MLV RNA by m5C, they do not rule out that virus propagation is affected, albeit in an indirect manner, by m5C changes. As a matter of fact, it has been repeatedly shown that interference with the m5C writer proteins (NSUN2, DNMT2) and/or m5C readers (ALYREF) perturb virus replication (Courtney et al. 2019a,b; Eckwahl et al. 2020). Thus, it is possible that the epitranscriptomic landscape of the host cell rather than that of the virus plays an important part in this context.
Conclusion
In this study, we have revisited the question of whether RNA viruses, such as SARS-CoV-2, HIV or MLV are subject to cytosine methylation. Using a rigorous bisulfite-sequencing protocol and stringent data analysis, we found no evidence for epitranscriptomic modification of viral RNA by cytosine methylation in any of the examined viruses. Our findings emphasize the necessity for the implementation of stringent experimental standards when studying m5C since all currently available methods for its detection (bisulfite-sequencing, m5C-immunoprecipitation, direct RNA sequencing using nanopore) are inherently error prone.
MATERIALS AND METHODS
Cells and virus infection
African green monkey Vero E6 cells stably overexpressing TMPRSS2 and ACE2 (Vero E6-TMPRSS2-ACE2) have been generated as described previously (Riepler et al. 2020) and were cultured in high glucose Dulbecco's Modified Eagle's Medium (Sigma) supplemented with 10% fetal calf serum (PAN), 2% L-Glutamin (Gibco), and 1% Penicillin-Streptomycin. To maintain expression of TMPRSS2 and ACE2, the cells were selected with Hygromycin (1:250) and Blasticidin (1:1000), respectively. 107 cells were seeded in a 175 cm² cell culture flask 1 d prior to infection. The cells were infected with a SARS-CoV-2 D614G isolate (B.1.177, GISAID: EPI_ISL_3305837) with an MOI of 0.001 and harvested after 48 h. PM1 cells (obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Drs. Paulo Lusso and Robert Gallo) were cultured in RPMI-1640 Medium (Sigma) supplemented with 5% fetal calf serum and 2% L-Glutamine (Gibco). 2 × 106 PM1 cells/well were seeded in a six-well plate and directly infected with replication competent HIV NL4-3 or HIV-1 D117/II isolate. Cells were harvested 48 h post infection. Mus dunni cells were cultured in RPMI-1640 Medium (Sigma) supplemented with 10% fetal calf serum and 2% L-Glutamine (Gibco). 1 × 106 cells/well were seeded 1 d prior to infection in a six-well plate. The cells were infected with an MOI of 0.1 Friend Murine Leukemia Virus (FMLV) and harvested 48 h post infection.
Bisulfite-sequencing (BS-seq)
Transcriptome-wide BS-seq
For transcriptome-wide analysis of SARS-CoV-2 methylation, poly(A) RNA was prepared from infected Vero E6-TMPRSS2-ACE2 cells using the NEBNext mRNA Magnetic Isolation Module (NEB), followed by two additional rounds of poly(A)RNA enrichment by Dynabeads (Ambion) according to the manufacturer's protocol. Bisulfite treatment was performed as described previously (Trixl et al. 2019) using the EZ RNA Methylation Kit (Zymo Research) with some modifications. Briefly, 0.7 µg of enriched poly(A)RNA (no prior fragmentation) were mixed with bisulfite solution and 20% formamide (Roth), and subjected to three cycles of incubation at 10 min 70°C and 45 min 64°C. Desulfonation was performed at 37°C for 20 min followed by purification according to the manufacturer's instructions. To each sample, 1 µL 1:10 diluted ERCC RNA Spike-In Mix (Thermo Fisher) was added before bisulfite treatment. The purified RNA was subjected to NGS library preparation using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) according to the manufacturer's recommendations except for the following modifications: (i) Because bisulfite treatment causes RNA fragmentation, no further fragmentation was performed. (ii) For first strand cDNA synthesis, the random primer mix supplied with the kit was supplemented with 1 µg custom made random hexamers lacking the C base. Next generation sequencing was performed on a HiSeq X instrument at ∼100 Mio 150 nt paired-end reads. Three biological replicates were processed.
BS-seq of PCR amplicons
Total RNA was extracted from virus-infected cells using TRIzol (Sigma-Aldrich) and digested with DNase I (NEB) for 20 min at 37°C followed by purification using phenol chloroform extraction and isopropanol precipitation. An amount of 1.2–1.5 µg total RNA was used for bisulfite treatment exactly as described above. cDNA synthesis was performed by the GoScript Reverse Transcription System (Promega) with the addition of 1 µg custom made random hexamers lacking the C base followed by PCR amplification using bisulfite compatible primers of the following genomic RNA regions: SARS-CoV-2 regions 28893–29051 and 29114–29274, HIV regions 2056–2200 and 9055–9256, MLV regions 303–468 and 6906–7040. Amplicons were cloned into pGEM-T vector (Promega) and plasmid DNA from individual colonies was analyzed by Sanger sequencing. To determine deamination efficiency, spike-in ERCC sequence 136 (negative control) and human tRNAAsp (positive control) were amplified and sequenced in parallel. Primer sequences are listed in Supplemental Table S2.
Bioinformatic analysis of BS-seq
Raw BS-seq reads were first cleaned from sequencing adapter contaminants using Trimgalore and aligned to the Chlorocebus sabaeus genome (Ensembl v106) extended with the SARS-CoV2 genome (ASM985889v3, Ensembl v101) and ERCC sequences using meRanGs from our previously published meRanTK software collection (Rieder et al. 2016). For m5C candidate detection, we extended the original version of meRanCall in the new release (meRanTK v1.3.0) to include a C-cutoff parameter, a signal-to-noise cutoff filter and a gene-specific methylation rate calculation. The following parameter settings were used: allowed duplicates <20 (-md 20), coverage >20 reads (-mcov 20), C-cutoff of 3 (-C_cutoff 3), signal-to-noise rate threshold 0.9 (-snr 0.9), methylation rate >0.2 (-mr 0.2). To determine statistical significance (FDR <0.01, -fdr 0.01) of the m5C calls, we used the gene-specific conversion rate (-gcr). In case the m5C position was not within gene regions defined in the GTF annotation files from Ensembl, we used the conversion rate determined by the unmethylated ERCC sequences.
For reanalysis of the published BS-seq data, MLV (GSE139606) and HIV (GSE157193) data sets were downloaded from the Gene Expression Omnibus (GEO) database and processed exactly as described above.
DATA DEPOSITION
BS-seq data have been deposited with the Short Read Archive (SRA) database under BioProject accession number PRJN908293.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank I. Delazer and R. Micura for critical reading and comments on the manuscript. This work was supported by the Austrian Science Fund (FWF) (P33936-B and SFB F8009-B to A.L.). Funding for open access charge was provided by the Austrian Science Fund (SFB F8009-B).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079549.122.
-
Freely available online through the RNA Open Access option.
- Received December 7, 2022.
- Accepted February 27, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Anming Huang is the first author of this paper, “No evidence for epitranscriptomic m5C modification of SARS-CoV-2, HIV and MLV viral RNA.” Anming is a post-doctoral fellow in the group of Alexandra Lusser at the Medical University of Innsbruck, where he also carried out his PhD. His research focuses on the distribution and function of m5C in mRNA.
What are the major results described in your paper and how do they impact this branch of the field?
SARS-CoV-2 has been causing serious negative impacts; scientists have been actively working on vaccine and therapeutic drug development and on the mechanism of viral infection. There have also been many studies on the characteristics of the virus itself. However, with regard to the modifications of viral RNA, due to the limitations of research techniques, it was still unclear whether SARS-CoV-2 viral RNA contains the epitranscriptomic 5-methylcytosine (m5C) modification. We applied a stringent bisulfite sequencing and bioinformatic data analysis workflow that included suitable filters to reduce the calling of false positive sites to prove that SARS-CoV-2 viral RNA does not contain m5C modification. We also reexamined reported m5C sites in HIV and MLV which we could not confirm. Our results show that research techniques related to the emerging area of RNA modifications need ongoing improvement in order to establish a robust foundation for future studies.
What led you to study RNA or this aspect of RNA science?
I am interested in all subjects within life sciences. I have studied veterinary medicine, aquatic animal immunology, developmental biology, epigenetics, etc. Relatively speaking, the most complicated subject is epigenetics, which is an important means of gene regulation. In many cases, it can be inherited and affected by environment; and the diversity and complexity of every cell and tissue in our human body are realized by the regulation of gene expression, so epigenetics is very important.
This discovery that histone modification can regulate gene activities marks the rise of epigenetics. About 15 years ago, scientists began to propose RNA epigenetics. In the development process, there have been many discoveries and breakthroughs, and continuous innovation of technology has brought us closer and closer to reality. At the same time, we must also maintain a skeptical attitude toward the results and conclusions presented during the development process. Only by starting from the most authentic data and knowledge can we uncover the deeper secrets of epigenetics step-by-step.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Since many data showed that m5C modification is detected on different viral RNAs, we believed that as well. So it came as a surprise when we were trying to confirm those high confidence m5C sites but did not succeed.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My supervisor, Professor Alexandra Lusser, gave us enough space to develop, but always provided support and help to ensure that we did not go in the wrong direction. This has been the case since I started my PhD studies in her group until now. In addition to getting good academic training, I have also learned many other skills from her; for example, she always asks many questions when we design our work, such as why we do this, which questions we want to answer, and is it convincing? If not, what to do, etc. After a series of questions for ourselves, a very comprehensive plan can be achieved. For me, she has had a profound, positive influence.
What are your subsequent near- or long-term career plans?
My basic scientific research experience and background in different disciplines make me want to continue my research in the RNA field, in particular by developing nucleic acid vaccines and drugs to prevent and treat animal infectious diseases. For the treatment of animal diseases caused by bacterial infections as well, I think this will certainly alleviate the threat of antibiotic resistance to the health of animals and humans.








