
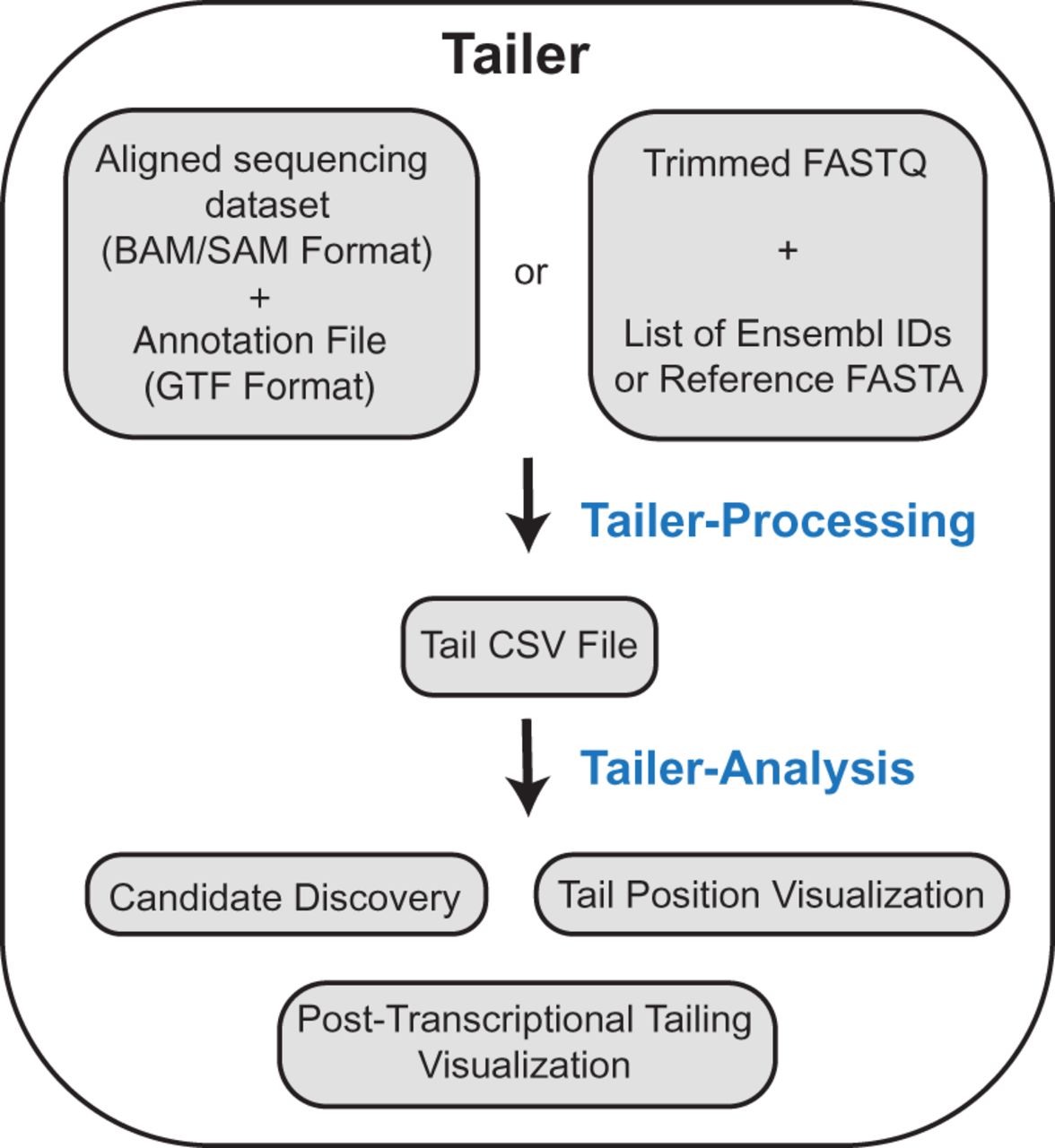
FIGURE 1.
A general overview of Tailer's workflow. Tailer is split into two major parts, a processing function, and an analysis web server. Tailer-processing infers RNA 3′ ends using a BAM/SAM alignment file and a GTF formatted annotation file, or a FASTQ sequence read file and a reference FASTA gene file (which can be generated from Ensembl IDs). For either method, the output is a standardized Tail CSV file, which can be analyzed directly, or fed into the Tailer-analysis web server for discovery of candidate tailing changes in comparison between data sets as well as visualization of tails with a variety of graphing tools.








