
Ribosome profiling reveals novel regulation of C9ORF72 GGGGCC repeat-containing RNA translation
- Heleen M. van ‘t Spijker1,4,
- Emily E. Stackpole1,4,
- Sandra Almeida2,4,
- Olga Katsara3,
- Botao Liu1,
- Kuang Shen1,
- Robert J. Schneider3,
- Fen-Biao Gao2 and
- Joel D. Richter1
- 1Program in Molecular Medicine, University of Massachusetts Chan Medical School, Worcester, Massachusetts 01605, USA
- 2Department of Neurology, University of Massachusetts Chan Medical School, Worcester, Massachusetts 01605, USA
- 3Department of Microbiology, New York University School of Medicine, New York, New York 10016, USA
- Corresponding authors: Joel.richter{at}umassmed.edu, Fen-biao.gao{at}umassmed.edu
-
↵4 These authors contributed equally to this work.
Abstract
GGGGCC (G4C2) repeat expansion in the first intron of C9ORF72 causes amyotrophic lateral sclerosis and frontotemporal dementia. Repeat-containing RNA is translated into dipeptide repeat (DPR) proteins, some of which are neurotoxic. Using dynamic ribosome profiling, we identified three translation initiation sites in the intron upstream of (G4C2) repeats; these sites are detected irrespective of the presence or absence of the repeats. During translocation, ribosomes appear to be stalled on the repeats. An AUG in the preceding C9ORF72 exon initiates a uORF that inhibits downstream translation. Polysome isolation indicates that unspliced (G4C2) repeat-containing RNA is a substrate for DPR protein synthesis. (G4C2) repeat-containing RNA translation is 5′ cap-independent but inhibited by the initiation factor DAP5, suggesting an interplay with uORF function. These results define novel translational mechanisms of expanded (G4C2) repeat-containing RNA in disease.
Keywords
INTRODUCTION
Expansion of GGGGCC (G4C2) repeats in C9ORF72 intron 1 is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The G4C2 repeat expansion may promote neurotoxicity through partial loss of the C9ORF72 protein, aggregation of the encoded repeat RNA, and/or accumulation of dipeptide repeat (DPR) proteins translated from both sense and antisense repeat RNA. Poly(GA), poly(GR), and poly(GP) are generated from the G4C2 sense strand and poly(PA), poly(PR), and poly(GP) from the antisense strand (Ash et al. 2013; Mori et al. 2013; Zu et al. 2013). Synthesis of DPR proteins has been proposed to occur by repeat-associated non-AUG (RAN) translation, where ribosomes directly associate with G4C2 repeat RNA and bypass 5′ cap-dependent 40S ribosomal subunit scanning (Zu et al. 2011). In support of this possibility, depletion of the cap-binding factor eIF4E has little effect on DPR protein synthesis in a reporter construct (Cheng et al. 2018). Mechanistically, it is unclear how such a direct ribosome–RNA interaction could occur, although ancillary proteins (Cheng et al. 2018) or a ribosomal protein (Yamada et al. 2019) may be involved. Studies based on an in vitro translation system primed with C9ORF72 pre-mRNA and reporter constructs indicate that poly(GR) and poly(GP) protein synthesis is promoted by a near-cognate CUG initiation codon in the GA frame followed by ribosome frameshifting on G4C2 RNA (Tabet et al. 2018). However, recent work indicates that intronic sequences 5′ to the repeat regulate poly(GR) synthesis (Lampasona et al. 2021).
To investigate the sites of translation initiation of C9ORF72 G4C2 repeat-containing RNA, we performed ribosome profiling after treatment with homoharringtonine (HHT), which “freezes” ribosomes on initiation codons while allowing elongating ribosomes to continue translocation (Ingolia et al. 2011; Lee et al. 2012; Shah et al. 2020). We applied this regimen to C9ORF72 patient-derived induced pluripotent stem cells (iPSCs) and neurons as well as HEK cells transfected with a reporter G4C2 repeat-containing construct, which allowed us to map ribosome initiation sites in the C9ORF72 intron upstream of G4C2 repeats. Ribosome footprints were detected at three sites irrespective of the presence of the repeats; two sites are in-frame with poly(GA) while another is in-frame with poly(GP). Several lines of evidence demonstrated that unspliced G4C2 repeat-containing mRNA is a primary substrate for DPR protein synthesis and that ribosomes may stall on the G4C2 repeats, which likely forms a hairpin structure (Wang et al. 2019). A uORF encoding a 6-kDa polypeptide conserved among primates beginning in exon 1 and extending into the intron represses downstream translation. Translation of G4C2 repeat-containing RNA does not require a 5′cap or cap-associated factors, indicating that 40S ribosomal subunit scanning is dispensable. These paradoxical results led us to consider atypical initiation promoted by death-associated protein 5 (DAP5), an eIF4G-like protein that regulates initiation independent of eIF4E through internal ribosome entry or cap-dependent translation using alternate cap-binding protein eIF3d (Marash et al. 2008; de la Parra et al. 2018; Haizel et al. 2020). Surprisingly, depletion of DAP5 promotes poly(GA)-frame intron translation, suggesting that it antagonizes uORF-mediated translation inhibition. Our results provide several new insights into DPR protein synthesis in C9ORF72-ALS/FTD.
RESULTS AND DISCUSSION
Ribosome profiling reveals initiation sites in a C9ORF72 intron
To identify translation initiation sites of C9ORF72 G4C2 repeat-containing RNA, we performed ribosome profiling on iPSCs derived from three patients with the expansion and three healthy individuals. iPSCs were treated with homoharringtonine (HHT) or cycloheximide (CHX). HHT freezes ribosomes on initiation codons but allows elongating ribosomes to continue translocation until they dissociate at a termination codon (Supplemental Fig. S1, i.e., ribosome run-off) (Ingolia et al. 2011; Lee et al. 2012); CHX freezes all ribosomes. Figure 1A shows that with the exception of the canonical C9ORF72 AUG initiation codon in exon 2, almost all ribosomes had run-off the RNA after HHT treatment; in contrast, ribosome footprints were readily detected on downstream coding exons after CHX treatment. We detected a very low level of ribosome footprints in intron 1 (Fig. 1A, red box) but not in other introns. Upon closer inspection of intron 1 (Fig. 1B), we found two clusters of footprints that were particularly evident in the C9ORF72 patient iPSCs treated with HHT (clusters 1 and 2). These clusters were also detected in patient cells treated with lactimidomycin D (LTM), which has an activity similar to HHT (Lee et al. 2012). Two-week-old cortical neurons differentiated from a C9ORF72 patient and control iPSC lines that were treated with HHT followed by ribosome profiling, displayed nearly the same two footprint clusters (Fig. 1C). The low footprint yield likely reflects that DPR proteins are expressed at very low levels in these cells. Further, the fact that the footprints are derived from an intron would also a priori lead to a low footprint yield. Codon resolution for the low number of footprints was unclear, which likely reflected the use of RNase A and T1 to generate the footprints as these enzymes do not digest RNA after adenosine residues. Despite these caveats, the observation of distinct footprint clustering in intron 1 suggests that they may represent start sites for G4C2 repeat-containing RNA translation. Total read counts for these experiments are provided in Supplemental Table 1.
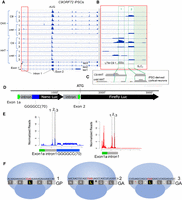
Ribosome profiling of C9ORF72. (A) iPSCs derived from three patients with expanded G4C2 repeats (C9) and three controls (cntrl) were treated with cycloheximide (CHX) or homoharringtonine (HHT) and then processed for ribosome profiling of the C9ORF72 RNA. Total read counts are provided in Supplemental Table 1. (B) An area of intron 1 upstream of G4C2 repeats was expanded to reveal two clusters of ribosome footprints. Also shown is an expanded area of intron 1 from a C9 patient following lactimidomycin D (LTM) treatment, which has an effect similar to HHT and ribosome profiling. (C) iPSC-derived neurons from a C9ORF72 patient and a healthy control were treated with HHT followed by ribosome profiling. The relevant area of intron 1 is shown. (D) A reporter construct containing a portion of C9ORF72 exon 1, C9ORF72 intron 1, G4C2(70), nano luciferase, a 3′ portion of C9ORF72 intron 1, the 5′ end of C9ORF72 exon 2, and firefly luciferase was expressed in HEK293T cells for ribosome profiling. (E) HEK cells were transfected with the plasmid noted above, or an identical one in which G4C2(70) was removed, followed by HHT treatment and ribosome profiling. Three ribosome footprints were detected on RNA derived from both plasmids, and are designated 1 (at 222), 2 (at 269), and 3 (at 281). The dip in the signal on the G4C2(70) RNA is due to a point mutation of an A for a G after 18 repeats in the reporter, which does not affect the reading frame. (F) Mapping of start sites with codon resolution shows that footprint 1 contains a putative CUG start site and is in-frame with poly(GP). Footprint 2 contains a putative CUG start site and is in-frame with poly(GA); however, a stop codon is immediately adjacent to this CUG codon. Footprint 3 contains a putative CUG start site in-frame with poly(GA).
To increase the sensitivity of the ribosome profiling, we used a previously described dual reporter plasmid (Cheng et al. 2018) containing portions of C9ORF72 exons 1 and 2 flanking intronic sequences that harbor 70 G4C2 repeats and nano luciferase; firefly luciferase is fused to exon 2 as an internal control (Fig. 1D). This reporter reflects patient neurons in that the repeats are properly located in the intron. Ribosome profiling was performed on HHT-treated HEK cells transfected with this plasmid or a control plasmid that did not contain G4C2(70). To ascertain codon resolution, we used RNase 1, which has no base preference. Figure 1E shows that three sets of footprints were detected in the intron preceding G4C2(70). Supplemental Figure S2 shows a comparison of the footprints generated by RNase A/T1 and a second replicate using RNase 1. RNase 1 hydrolyzes RNA adjacent to the ribosome, allowing us to identify 29-nt mRNA fragments with codon resolution (Supplemental Fig. S3). Initiation site mapping (Ingolia et al. 2009; Lee et al. 2012) identified footprint 1 (CUG at nucleotide 222) that could initiate in the poly(GP) frame but is also in-frame with a subsequent termination codon directly preceding the repeat expansion (Fig. 1F). Footprint 2 (CUG at nucleotide 269) was in the poly(GA) frame but immediately adjacent to a termination codon. Footprint 3 (CUG at nucleotide 281) is in the poly(GA) frame with no intervening stop codon prior to G4C2(70), suggesting that footprint 3 is likely to translate poly(GA) RNA. Also note that although the canonical start codon for C9ORF72 protein is in exon 2, there were putative initiating ribosome footprints in exon 1. Importantly, Figure 1E also shows that the same three ribosome footprints were detected when G4C2(70) is not present, thus repeat expansion does not position ribosomes in the intron. These data indicate that in healthy individuals without G4C2 repeat expansion, C9ORF72 intron 1 RNA is likely to be translated as well.
Poly(GA) and poly(GP)-frame initiation
We mutated the putative start codons identified by ribosome profiling and assessed the outcomes on translational efficiency by measuring luciferase activity relative to RNA levels. Figure 2A shows that changing the CUG to CCG in footprint 3 inhibited poly(GA) frame translation as assessed by nano luciferase activity, but had no significant effect on firefly luciferase activity. Firefly RNA, which is mostly spliced, serves as an internal control and is likely translated in a “conventional” manner. This alteration significantly increased nano activity in the poly(GP) frame but not firefly luciferase activity. These data indicate that the CUG in ribosome footprint 3 is the start site for poly(GA) frame translation, as has been noted by others (Green et al. 2017; Sonobe et al. 2018; Tabet et al. 2018; Almeida et al. 2019; Lampasona et al. 2021). The observation that mutation of this CUG also up-regulates poly(GP) frame nano translation is paradoxical but could be explained if the reading frame was shifted within the G4C2 repeat (Tabet et al. 2018). We could detect only a very low amount of translation in the poly(GR) frame under the same conditions as the other two reading frames and thus it was not pursued further. However, intronic sequence requirements for poly(GR)-frame translation have been examined (Lampasona et al. 2021).
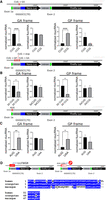
Functional analysis of translation start sites based on ribosome profiling. (A) The CUG in ribosome footprint 3 was mutated to CCG and the plasmid was transfected into HEK cells for 24 h. Nano and firefly luciferase activities and nano and firefly RNA levels (by RT-qPCR) in the poly(GA) and poly(GP) frames were measured and their relative translational activities (activity to RNA) were plotted. (B) The CUGs in the three ribosome footprints were mutated to CCG and nano and firefly activities and their RNAs in the poly(GA) and poly(GP) frames were measured and plotted as above. (C) The AUG at the end of exon 1 was mutated to UAA and nano and firefly luciferase activities in the poly(GA) and poly(GP) frames were measured and plotted as above. (*) P < 0.05; (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001 (Student's t-test). (D) uORF formation and alignment of amino acid sequences of the uORF in human, orangutan, and macaque.
Simultaneous mutation of the CUGs in the three ribosome footprints elicited a decrease in poly(GA) frame translation as expected, but also a decrease in poly(GP) frame translation (Fig. 2B), indicating that footprint 1 likely harbors the start codon for the poly(GP) frame. We surmise that translocating ribosomes starting from this CUG read through the stop codon immediately preceding G4C2(70) in this frame.
A conserved uORF initiating in exon 1
We also identified ribosome footprints on an AUG near the end of exon 1 (although the footprints from the reporter do not precisely correlate with the AUG at the end of exon 1; cf. Fig. 1E), which has been suggested to be the start site of a uORF (Tabet et al. 2018). Indeed, mutation of this site increased poly(GA) and poly(GP)-frame translation (Fig. 2C). Moreover, using mass spectrometry, we identified a peptide derived from this uORF that is encoded by intron sequences (Supplemental Fig. S4A); this peptide was not detected when the exon 1 AUG was changed to UAA (Fig. 2D). Interestingly, this 180 base uORF encodes a ∼6 kDa polypeptide, which is unusually large in that only <5% of uORFs are at least this size (Rodriguez et al. 2019). The amino acid sequence of the uORF is conserved among higher primates, which may indicate that it has a biological function (Fig. 2D). The uORF is predicted to form a helix–loop–helix (HLH) configuration (Supplemental Fig. S4B,C; Yang and Zhang 2015) but lacks basic residues at the amino terminus that is typical of transcription factors (Murre 2019). However, small HLH peptides that inhibit protein–protein interactions have been developed (Mudiyanselage et al. 2020) and the naturally occurring small HLH Id proteins, which are 134–178 amino acids in length in humans, perform a variety of biological tasks by interfering with transcription factors (Ruzinova and Benezra 2003). Although we have not assessed a biological activity of the C9ORF72 uORF polypeptide, it may have a function in cells that have strong C9ORF72 promoter activity. The nucleotide sequence of the uORF is even more highly conserved (Supplemental Fig. S4D) throughout mammals. By way of comparison, introns 2 and 3 of C9ORF72 have no detectable conservation at either the amino acid or nucleotide sequence levels.
The observation that the exon 1 AUG alters intronic DPR protein synthesis suggested that unspliced RNA is a substrate for G4C2 repeat-containing RNA translation. To test this, we pelleted polysomes from transfected HeLa Flp-In cells by ultracentrifugation and determined the relative amount of RNA in this fraction by RT-qPCR using primers that are complementary to exon 1 and intron 1 (primer pair 1), intron 1 only (primer pair 2), and intron 1 with exon 2 (primer pair 3); EDTA release of polysomes served as the control (Fig. 3A). Spliced C9ORF72 RNA from exon 1 to exon 2 was also measured and was more abundant than unspliced RNA from intron 1 to exon 2 (primer pair 3, measured relative to hygromycin RNA, which is expressed from the C9ORF72 reporter plasmid) (Fig. 3B). These results indicate that the majority of the repeat-containing RNA is spliced, consistent with previous reports (Tran et al. 2015; Cheng et al. 2018). The pelleted polysome fractions contained significant amounts of spliced and unspliced RNA as deduced with each primer pair (Fig. 3C). Irrespective of the primer pairs used to measure RNA, the amount of transcript in the polysome pellet was nearly identical; and indeed, the ratios of RT-qPCR products using exon 1 and intron 1 primers compared to intron 1 primers only were both 1 (Fig. 3D). These data were confirmed in C9 iPSCs (as in Fig. 1A) that express endogenous C9ORF72 and DPR proteins (Fig. 3E,F). Therefore, unspliced RNA is a primary substrate for DPR protein synthesis, which is concordant with other studies that assessed endogenous C9ORF72 splicing (Niblock et al. 2016; Sznajder et al. 2018).
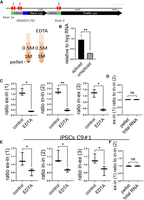
Substrate for C9ORF72 intron translation. (A) Lysates from HeLa Flp-In cells stably expressing the reporter plasmid were ultracentrifuged through 0.5 M and 1 M sucrose to pellet polysomes. Parallel experiments were performed when lysates were treated with EDTA, which dissociated polysomes. (B) Spliced RNA was measured from exon 1 to exon 2 and was far more abundant in our input compared to unspliced RNA (pair 3), relative to hygromycin (hyg) RNA expressed from the same plasmid. (C) RNA from the pellets as well as input was subjected to RT-qPCR with the primers in intron 1 and exon 1 (pair 1), within intron 1 (pair 2), and intron 1 and exon 2 (pair 3). (D) The ratios of the RT-qPCR products using primer pair 1 (exon 1 and intron 1) to primer pair 2 (both in intron 1) in the polysome pellet and total input RNA is shown. (E) Lysates from iPSCs C9#1 (also referred to as C9 26#6) were ultracentrifuged through 0.5 M and 1 M sucrose to pellet polysomes. RNA from the pellets was subjected to RT-qPCR with the primers in intron 1 and exon 1 (pair 1), within intron 1 (pair 2), and intron 1 and exon 2 (pair 3). Parallel experiments were performed when lysates were treated with EDTA, which dissociated polysomes. (F) The ratios of the RT-qPCR products using primer pair 1 (exon 1 and intron 1) to primer pair 2 (both in intron 1) in the polysome pellet and total input RNA are shown. All experiments were performed in biological triplicate. (*) P < 0.01, (***) P < 0.001 (Student t-test).
DAP5 regulation of G4C2 repeat-containing RNA translation
To ascertain the mechanism of translation initiation on the G4C2 repeat-containing RNA, we focused on the poly(GA) frame and placed a strong hairpin (−50 kcal/mol) at the 5′ end of exon 1, which would prevent cap-dependent 40S ribosome subunit binding and 5′ UTR scanning (Babendure et al. 2006). This structure had no significant effect on poly(GA)-frame nano luciferase translation relative to an unstructured sequence but strongly reduced firefly luciferase translation, which would be expected if translation of this spliced RNA was initiated from the cap (Fig. 4A). We also treated transfected HEK cells with 4EGI, a small molecule that disrupts eIF4E–eIF4G and inhibits eIF4E-mediated cap-dependent translation (Moerke et al. 2007) and thereby impairs higher cognitive function (Hoeffer et al. 2011). Subsequent treatment with HHT and ribosome profiling shows that this regimen had no effect on the three ribosome footprints in intron 1 (Fig. 4B), again indicating that translation of G4C2 repeat-containing RNA is independent of cap-binding by either eIF4E or eIF3d.
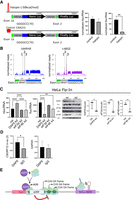
Mechanism of C9ORF72 intron translation. (A) A hairpin (−50 kcal/mol) or an identical size control sequence CAA(21) that forms no hairpin was inserted two bases downstream from the cap in the reporter construct. The plasmids were transfected into HEK cells followed by determination of nano and firefly luciferase activities and RNA levels. (B) The constructs shown in Figure 1D were transfected into HEK cells followed by HHT treatment as well as 4EGI, a small molecule inhibitor of the eIF4E–eIF4G interaction. 4EGI was applied to cells at 100 µM for 3 h. Ribosome profiling was then performed as in Figure 1E. (C) A HeLa Flp-In cell line expressing the construct depicted in Figure 1D was transduced with lentiviruses expressing a nonspecific sequence, negative siRNA (nsi) or shRNAs for DAP5, eIF3d, or eIF4E. Nano and firefly luciferase activities and RNAs were determined. These proteins as well as vinculin were then western blotted and quantified. (D) C9ORF72 and GAPDH RNA immunoprecipitation was performed with DAP5 antibody or nonspecific IgG on HeLa Flp-In cells. RNA values were normalized to DAP5, which was set at 1. All experiments were performed in biological triplicate. (*) P < 0.01, (***) P < 0.001 (one-way ANOVA). (E) A model for C9ORF72 GGGGCC repeat-containing RNA translation. Ribosome profiling shows initiating ribosomes on 3 CUG codons upstream in the intron of G4C2(n). Two of the CUG codons are in-frame with poly(GA) and one in-frame with poly(GP). A uORF initiating at an AUG codon at the end of exon 1 is a negative regulator of translation. Recruitment or positioning of ribosomes on the uORF AUG may be facilitated by the eIF4G-like protein DAP5. Ribosome stalling may occur on the G4C2(n) hairpins formed by the repeat expansion. Created with BioRender.com.
Thus far, we have shown that (i) ribosome positioning in intron 1 does not require expanded G4C2 repeats, (ii) DPR protein synthesis takes place mostly on unspliced RNA, (iii) ribosomes may be stalled on expanded G4C2 repeats, (iv) intron translation is regulated by a uORF conserved among primates, and (v) translation of expanded G4C2 repeats occurs in a cap-independent manner (Fig. 4A,B). These data suggest that an atypical translation factor(s) may be involved in driving DPR protein synthesis. One such factor is death-associated protein 5 (DAP5), an eIF4G-like protein that lacks eIF4E and poly(A) binding protein domains (Imataka et al. 1997; Alard et al. 2019). DAP5 regulates the translation of many mRNAs (Lee and McCormick 2006; de la Parra et al. 2018) by either recruiting ribosomes to structured internal ribosome entry sites (IRES) (Liberman et al. 2015) or 5′ untranslated regions (UTRs) (Haizel et al. 2020), or by cap-dependent mRNA translation as a complex with eIF3d (de la Parra et al. 2018). Depletion of DAP5 increased translation of poly(GA)-frame nano luciferase; depletion of eIF3d or eIF4E had no effect (Fig. 4C). DAP5 as well as eIF4E depletion had a strong inhibitory effect on firefly luciferase translation (Fig. 4C). Moreover, C9ORF72 RNA coimmunoprecipitated with DAP5 antibody relative to IgG control (Fig. 4D). Control GAPDH RNA coimmunoprecipitation with DAP5 antibody or IgG control was indistinguishable. To account for the DAP5 inhibitory effect on G4C2(70) translation, we propose a model in which this factor recruits ribosomes to the exon 1 uORF AUG, in this manner, both DAP5 and uORF translation are negative regulators of DPR protein synthesis (Fig. 4E). Initiation with DAP5 can be cap-independent (de la Parra et al. 2018), which could explain why the hairpin structure does not affect initiation at the uORF AUG (Fig. 4A).
Our results do not preclude the involvement of other factors such as DDX3x (Cheng et al. 2020) or RPS25 (Yamada et al. 2019) in DPR protein production. In fact, DDX3x was found to be a strong binding partner of DAP5 by protein–protein mass spectrometry analysis (de la Parra et al. 2018). Motor neurons may be particularly sensitive to the levels of such ancillary proteins, which could, for example, result in higher levels of G4C2(n) repeat translation. Ribosome profiling of C9ORF72 in motor neurons and examination of levels of DAP5 and other factors in these cells would be important future steps in assessing translational regulation of expanded G4C2 repeat RNA.
Repeat-associated non-AUG (RAN) translation was originally envisioned as a process where ribosomes initiate translation through direct association with expanded microsatellite repeat sequences (Zu et al. 2011). Tabet et al. (2018) suggested that the G4C2 repeat expansion binds individual ribosomal subunits that have no translational capacity. In contrast, our ribosome profiling data show that full 80S ribosomes are associated with G4C2 repeats even after HHT treatment and ribosome run-off. A possible explanation is that these ribosomes are stalled on the repeat after initiating upstream (Fig. 4E). Ribosome stalling, which cannot be revealed by “conventional” ribosome profiling or ribo-tag pulldown experiments (Richter and Zhao 2021), occurs on thousands of RNAs in the brain (Shah et al. 2020). Although ribosomes progressively unwind RNA as they translocate to form the DPR proteins, they can be directly or indirectly be impeded by proteins such as FMRP (Shah et al. 2020), polyproline tracts in nascent peptides (Pavlov et al. 2009), or phosphorylation of eukaryotic elongation factor 2 (eEF2) (Dever and Green 2012). The hairpin structure likely formed by expanded G4C2 repeats in C9ORF72 (Wang et al. 2019) is almost certainly sufficient to slow ribosome translocation. Moreover, poly(GR) may slow ribosome translocation as well (Loveland et al. 2020). Using an in vitro RNA folding model, Wang et al. (2019) calculated that six G4C2 repeats have a ΔG of −22 to −16 kcal/mol. Given that translocating 80S ribosomes generate ∼3.8 kcal/mol of energy (Liu et al. 2014), they could slow or stall on the many hairpin structures that would be formed on an endogenous G4C2 expansion. Ribosome stalling on the expanded repeat would suggest a prolonged period of time would be required to produce and accumulate neurotoxic DPR proteins, which could be partly responsible for the late onset of ALS and FTD.
MATERIALS AND METHODS
Cell lines
HEK293T was purchased from ATCC. HeLa Flp-In cells were donated by Dr. Shuying Sun (Johns Hopkins Medical School), and expression of the heterologous C9ORF72 reporter was induced with doxycyclin.
iPSC lines
All iPSC lines used in this study were previously generated and characterized: control male 2#20 in Almeida et al. (2012); control female 37#20 in Zhang et al. (2013); control male 35#11 in Freibaum et al. (2015); C9ORF72 mutation carrier female 26#6 and male 27#11 in Almeida et al. (2013); C9ORF72 mutation carrier male 29ALS in Sareen et al. (2013). The C9ORF72 mutation carrier lines recently underwent authentication by short tandem repeat (STR) analysis (Lopez-Gonzalez et al. 2019).
Ribosome profiling of HEK293T cells
For experiments in HEK293T cells, the C9ORF72-GA or C9ORF72-Neg-GA reporters were first transfected into HEK293T cells using Lipofectamine2000 (Thermo Fisher) following the manufacturer's guidelines. Briefly, 22 µg of plasmid DNA was transfected into HEK293T cells plated on 15-cm dishes at a ratio of 1:2.5 DNA:Lipofectamine. Twenty-four hours post-transfection, HEK293T cells were treated with either homoharringtonine (LKT Laboratories) or cycloheximide (Sigma). For homoharringtonine treatment, homoharringtonine was added to the medium for a final concentration of 2 µg/mL and incubated at 37°C for 5 min. Cycloheximide was subsequently added to the media (100 µg/mL) and the cells were then washed twice with ice-cold PBS containing cycloheximide (100 µg/mL). Cells were then collected by scraping, pelleted by centrifugation, snap-frozen in liquid nitrogen, and immediately stored at −80°C. For cycloheximide treatment alone, cycloheximide was added to the medium for a final concentration of 100 µg/mL for a 1 min incubation. Cells were then washed, pelleted, and snap-frozen as stated above. Two HEK293T replicates were used for our analyses.
Cell pellets were thawed for 5 min in ice-cold polysome lysis buffer (20 mM Tris pH 7.4, 5 mM MgCl2, 100 mM KCl, 1% NP40, 1× EDTA-free protease inhibitor cocktail (Roche), 1 mM DTT (Thermo Fisher), 25 U/mL Turbo DNase I (Thermo Fisher), and 100 µg/mL cycloheximide in nuclease-free water). Cell pellets were then gently dissociated by pipetting, incubated on ice for a further 10 min, and lysed by trituration through a 25-G needle 10 times. After a 5 min incubation on ice, lysates were clarified by centrifugation at 10,000g for 10 min at 4°C. Supernatants were collected and the amounts of nucleic acids (A260) were determined by NanoDrop. For each sample, 20%–25% of cytoplasmic lysate had RNA extracted using TRIzol LS reagent for RNA-seq.
For RNase I digestion, we followed a published protocol (Lee et al. 2012). Lysates were first layered on 10%–50% sucrose gradients prepared in polysomes buffer (20 mM Tris pH 7.4, 5 mM MgCl2, 100 mM KCl, 1 mM DTT, and 100 µg/mL cycloheximide) and ultracentrifuged in a SW41Ti rotor (Beckman Coulter) at 35,000 rpm for 2.5 h at 4°C. Gradients were fractionated at 1.5 mL/min and a 20 sec collection interval using a Brandel fractionation system that continually monitored A260 values. The monosome through polysome fractions was identified and pooled to a volume of 140 µL. RNase I (2 µL; Ambion) was added directly to this pool and incubated at 4°C for 1 h with end-over-end rotation. After digestion was complete, RNA was extracted using TRIzol LS following the manufacturer's protocol.
Cells were digested with RNase A/TI digestion following a published protocol (Heyer et al. 2015; Liu et al. 2018). Briefly, lysates were digested for 45 min at 25°C with 100 ng RNase A (Sigma #R4875) and 60U RNase TI (Thermo Fisher) per A260. The digestion was then stopped by the addition of 2.5-µL SUPERase In RNase Inhibitor (Ambion) and placed on ice. Digested lysates were loaded onto 10%–50% sucrose gradients prepared in polysome buffer and ultracentrifuged as stated above. Gradients were then fractionated at 1.5 mL/min and a 12 sec collection interval using the Brandel fractionation system. Fractions containing the monosome were identified, pooled, and extracted with TRIzol LS according to the manufacturer's protocol.
Ribosome profiling of iPSCs
Cell pellets were thawed and lysed as described above for HEK293T cells. Supernatants were collected, and the amounts of nucleic acids (A260) were determined by NanoDrop. Cell lysates were digested with RNase A/TI digestion exactly as described above for HEK293T cells (Heyer et al. 2015; Liu et al. 2018). After digestion, 2.5µL SUPERase In RNase Inhibitor (Ambion) was added and lysates were placed on ice. Digested lysates were loaded onto 10%–50% sucrose gradients prepared in polysome buffer and ultracentrifuged as stated above. Gradients were then fractionated as stated above and monosome fractions were identified, pooled, and extracted with TRIzol LS according to the manufacturer's protocol.
Ribosome profiling of iPSC-derived neurons
Cell pellets were thawed and lysed as described above for HEK293T cells. Supernatants were collected and the amount of RNA was determined using the Qubit High-Sensitivity RNA Kit. For RNase A/TI digestion, we followed a previously published protocol (Liu et al. 2019) where the RNase A and T1 amounts are kept constant for low-input samples. Lysate was digested with 1242 ng RNase A (Ambion #AM2270) and 155U RNase TI (Thermo Fisher Scientific #EN0542) for 30 min at 25°C (concentration #4 from Liu et al. 2019). Digested lysates were loaded onto 10%–50% sucrose gradients prepared in polysome buffer and ultracentrifuged as stated above. Gradients were then fractionated at 1.5 mL/min and a 12 sec collection interval using the Brandel fractionation system. Fractions containing the monosome were identified, pooled, and extracted with TRIzol LS according to the manufacturer's protocol.
Ribosome profiling library preparation
Ribosome profiling libraries were prepared following published protocols (Heyer et al. 2015; Liu et al. 2018, 2019). First, rRNA was depleted from the purified monosomal RNA samples using RiboZero Gold (Illumina) and the remaining RNA samples were separated on a 15% TBU gel (National Diagnostics). Ribosome footprints were size-selected using 26–34 nt markers. Gel pieces were crushed and RNA eluted in RNA elution buffer (300 mM NaOAc pH 5.5, 1 mM EDTA, 0.25% SDS in nuclease-free water) overnight at room temperature. Gel pieces were filtered out by Spin-X centrifuge tube filters (Corning) and RNA precipitated overnight with equal volumes of isopropanol. Recovered RNA was dephosphorylated using T4 Polynucleotide Kinase (NEB) and then ligated to a preadenylated adaptor in miRCat-33 conversion oligos pack (IDT) using T4RNL2Tr.K227Q ligase (NEB). RNA was reverse transcribed using primers containing 5 nt-barcodes and 8 nt-unique molecular identifiers (UMIs) along with SuperScript III and first-strand buffer without MgCl2 (50 mM Tris–HCl pH 8.3, 75 mM KCl). Any remaining RNA was hydrolyzed by incubation with NaOH, RT products were precipitated overnight and separated on a 10% TBU gel. The 130–140 nt region was selected and gel pieces were crushed and cDNA eluted overnight at room temperature using DNA elution buffer (10 mM Tris pH 8.0, 300 mM NaCl, 1 mM EDTA in nuclease-free water). Gel pieces were filtered out by Spin-X centrifuge tube filters and cDNA precipitated overnight using an equal volume of isopropanol. Recovered cDNA was circularized with CircLigase (Epicentre). For samples digested with RNase A/TI, an additional round of rRNA depletion was performed by hybridizing cDNA derived from remaining rRNA to biotin-labeled antisense probes (IDT) and removal with Dynabeads MyOne Streptavidin C1 (Thermo Fisher). The optimal PCR cycle was first determined empirically by test PCR using the KAPA Library Amplification Kit (Kapa Biosystems) with titrated cycle number. The samples were then PCR amplified, separated on an 8% TBE gel, and the 180–190 nt products were selected. Gel pieces were crushed, eluted overnight at room temperature in DNA elution buffer, filtered, and precipitated overnight with equal volumes of isopropanol. The final DNA libraries were purified using AMPure XP beads (Beckman Coulter) following the manufacturer's guidelines. The final concentration of each library was determined using a KAPA Library Quantification Kit (KAPA Biosystems). Libraries were then pooled, denatured, diluted, and sequenced on a NextSeq 500 sequencer (Illumina) using a NextSeq 500/550 High Output Kit (Illumina) for 75 bp single-end runs.
Read processing and mapping
Individual samples were first separated based on the 5-nt barcode sequences and then adaptor sequences were removed using cutadapt. Trimmed reads were quality filtered and mapped to human rRNA and tRNA references using Bowtie2. Tophat2 was used to then map the remaining unmapped reads to a modified hg19 human genome containing the C9ORF72 reporter sequence. PCR duplicates were marked based on the UMI sequences and removed. Due to our modified genome containing both endogenous C9ORF72 and reporter C9ORF72 reporter gene sequences, multimapped reads were included to retain reads mapping to sequences shared between these two genes (e.g., exon 1a, exon 2, and intronic sequences 5′ to the G4C2 repeat).
The RPF length distributions, P-site offsets, and frame preferences were calculated using Plastid and Ribo-TISH (Dunn and Weissman 2016; Zhang et al. 2017). For HEK293T samples treated with RNase I, reads either (1) 29 and 30 nt in length or (2) all lengths were counted at every nucleotide position of the C9ORF72 reporter transcript by using the 13th nucleotide of the read for the P-site position. For HEK293T samples treated with RNase A/TI, reads either (1) 30 and 31 nt in length or (2) all lengths were counted at each nucleotide position using the 13th nucleotide of the read for the P-site position. See figure legends for which read length was used for individual analyses. For read aggregation plots, the number of RPF reads aligned to each position of the C9ORF72 transcript was normalized to both library depth and RNA level.
RNA-seq library preparation, read processing, and mapping
RNA-seq libraries were prepared from RNA isolated from cytoplasmic lysates (supernatants) generated during cell lysis for ribosome profiling. RNA (2.5–5 µg) first underwent poly(A)-selection using NEXTFLEX poly(A) beads following the manufacturer's instructions. Poly(A)-selected mRNA was then used to generate RNA-seq libraries using the NEXTFLEX Directional qRNA-Seq Kit according to the manufacturer's instructions. The final concentration of each library was determined using a KAPA Library Quantification Kit (KAPA Biosystems). Libraries were then pooled, denatured, diluted, and sequenced on a NextSeq 500 sequencer (Illumina) using a NextSeq 500/550 High Output Kit (Illumina) for 150 bp paired-end runs. For HEK293T samples harboring the C9ORF72 reporters, sequences were mapped to our modified hg19 human transcriptome containing the C9ORF72 reporter sequence using RSEM. For iPSC samples, sequences were mapped to the hg19 human transcriptome.
Cell culture
iPSC lines from three C9ORF72 carriers and three healthy individuals were cultured on Matrigel and mTeSR1. Cortical neuron differentiation was performed as described (Almeida et al. 2013). HEK293T cells cultured in DMEM with 20%FBS and antibiotic–antimyotic were transfected with reporter plasmids using lipofectamine 2000. HeLa Flp-In cells (donated by Dr. Shuying Sun, Johns Hopkins Medical School) were cultured in DMEM with 10%FBS and pen/strep and expression of the heterologous C9ORF72 reporter was induced with doxycyclin. DAP5, eIF3d, or eIF4E were depleted by lentiviruses expressing shRNA (de la Parra et al. 2018). Knockdown was confirmed by western blotting.
Lentivirus
Lentivirus was produced in HEK293T cells by cotransfection of psPAX2, pMD2, and short hairpin plasmid with lipofectmine 3000. The medium was changed after 24 h and the virus was collected 48 and 72 h after infection. The virus was filtered on 45-µm filters, concentrated on Amicon Ultra filter units, and titers measured using the ABM qPCR Lentivirus Titration Kit according to the manufacturer's instructions. DAP5, eIF3d, or eIF4E were depleted by lentiviruses expressing shRNA (de la Parra et al. 2018) and knockdown was confirmed by western blotting.
Luciferase assays
HEK293T cells were transfected with the C9ORF72 reporter with Lipofectamine 2000 and luciferase assays performed with the NanoGlo Dual Luciferase Reporter Assay System (Promega) according to manufacturer's instructions. Briefly, 24 h after transfection cells were lysed in passive lysis buffer (Promega) for 15 min at room temperature after which samples were collected and 20 µL of each sample was added to a Corning 96-Well clear bottom black microplate (Costar). An amount of 50 µL of ONE-glo reagent was added to each well and firefly luciferase was measured on a Tecan satire2 plate reader. An amount of 50 µL of STOP reagent was added to each well and nano luciferase was then determined; results were analyzed with Prism software.
Cloning
Construction of the bicistronic, spliceable C9ORF72 reporter plasmids harboring no repeats or ∼70 G4C2 repeats in either the GA and GP frames has been previously described (pcDNA5-FRT-TO-C9orf72; Cheng et al. 2018) (donated by Dr. Shuying Sun, Johns Hopkins Medical School). We modified these reporters to harbor ∼70 G4C2 repeats in the GR frame by inserting +2 “G” nucleotides during PCR amplification of the Nluc gene. As the no-repeat control from Cheng et al. (2018) only contains a partial region of the C9ORF72 intron 5′ to the G4C2 repeats, we generated a new no-repeat control plasmid that contained the entire 5′ intron sequence by first digesting pcDNA5-FRT-TO-C9ORF72-GA with BssHII and EcoNI and then inserting in oligonucleotides containing the following sequence: 5′-CGCGCTAGGCGGCCGCGTCTTCACACTCGAAGATTTCGTTGGGGACTGGCGACAGACAGCCGGCTACAACCTGGACCAAGTCCTTG-3′. Different reading frames of this no-repeat control was achieved by inserting +1 or +2 “G” nucleotides during PCR amplification of the Nluc gene. To generate the point mutation constructs #1 and #2 (CTG > CCG), we digested the C9ORF72 reporter plasmids with HindIII and BssHII and inserted phosphorylated oligonucleotides containing the different point mutations (New England Biolabs; Table 1). All oligonucleotides were designed to continue harboring HindIII and BssHII sites in the reporter. To generate the point mutation construct #3 (ATG > TAA), we digested with NheI and HindIII and inserted phosphorylated oligonucleotides containing the mutation (New England Biolabs; Table 1). For the constructs containing a −50.2 kcal/mol hairpin or control hairpin at the 5′ cap, we followed a previously published design (Babendure et al. 2006). Briefly, reporter constructs were digested with SacI and phosphorylated oligonucleotides were inserted that contained either the −50.2 kcal/mol hairpin (CCCTGCGGTCCACCACGGCCGATATCACGGCCGTGGTGGACCGCAGGG) or a control sequence (CAA21) at the +1 position.
Oligonucleotides for cloning
RT-PCR
RNA was isolated using TRIzol LS (Thermo Fisher) according to the manufacturer's instructions. Turbo DNAse treatment to remove genomic DNA was performed for 30 min at 37°C with constant shaking. Reverse transcriptase was performed with the Quantitect Reverse Transcription Kit on 1 µg RNA per sample according to manufacturer's guidelines. In short, after gDNA was removed, cDNA was produced with reverse transcription at 42°C followed by inactivation at 95°C. Specific primers pairs were designed for exon 1-intron 1 intron 1-intron 1 and intron 1-exon 2, firefly luciferase, nano luciferase, and hygroB. iTaq Universal SYBR Green Supermix was used to run RT-PCR on a QuantStudio 3 system with a Comparative Ct program and analyzed in QuantStudio design and analysis software v1.5.1. Expression values (ΔCT) were normalized to Hygromycin B mRNA (see Table 2).
Sequencing primers
Polysome pellets
HeLa Flp-In cells were stimulated with doxycycline and grown on 10-cm plates until confluent. C9 iPSC line 26#6 was grown as described previously on 15-cm plates until confluent. Cells were treated with 100 µg/mL cycloheximide (CHX) for 1 min after which they were centrifuged for 10 min at 2000g and then at 20,000g for 10 min at 4°C. The supernatants were centrifuged at 80,000 rpm for 3 h through 0.5 M and 1 M sucrose in polysome lysis buffer (20 mM Tris–HCl pH 7.4, 100 mM KCl, 5 mM MgCl2 100 µg/mL CHX, 1 mM DTT, 1× EDTA-free protease inhibitor, and 0.3% Triton-X100). The RNA was purified with TRIzol LS and quantified by RT-qPCR.
Mass spectrometry
HEK293T cells were grown on 10-cm plates till 70% confluent. Cells were transfected with 5 µg of plasmid with lipofectamine 2000, followed by a medium change after 24 h. The samples were collected in lysis buffer (150 mM NaCl, 1% NP40, 50 mM Tris pH 8), incubated on ice for 10 min followed by centrifugation at 12,000 rpm for 20 min at 4°C. The supernatants were collected, applied to an SDS gel, and the region below 10 kDa was digested in-gel with trypsin. NanoLC–MS/MS analysis was performed on the Orbitrap Lumos mass spectrometer. Data analysis was performed against the Swissprot human database appended with the uORF protein sequence with the Mascot search engine. Further annotation was performed in Scaffold.
RNA immunoprecipitation
DAP5 antibody (Cell Signaling Technologies) and IgG control were bound to radioimmunoprecipitation assay (RIPA)-buffer-washed Protein A/G magnetic beads (invitrogen). Cells were washed with ice-cold PBS and fixed with 1% formaldehyde in PBS for 10 min at 25°C with gentle rocking. Formaldehyde was quenched by adding glycine to a final concentration of 0.25 M and then incubated at 25°C for 5 min. Fixed cells were washed three times with ice-cold PBS and resuspended in 0.5 mL of RIPA-buffer (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1% Igepal CA-630, 0.1% SDS, 0.5% sodium deoxycholate) with protease inhibitors (Roche) and 1 mM DTT per three million cells. DNA was sheared by sonication on the ice twice at 15% amplitude for 2 sec ON, 10 sec OFF for a total of 30 sec. Lysates were incubated on ice for 10 min. Digest with Turbo DNase I and partial digestion with RNase I was performed for 3 min at 37°C with mixing (2 µL Turbo DNase I and 5 µL of 1–25 times diluted RNase I in PBS per 0.5 mL of lysate). Tubes were immediately transferred to ice and incubated for 5 min. Lysates were centrifuged at 21,000g at 4°C for 10 min. An amount of 200–500 µg of protein was supplemented with SUPERase In RNase Inhibitor (100 U mL−1, Ambion). The lysate was added to antibody-bound beads and incubated at 4°C for 12 h. Beads were washed five times with 500 µL RIPA buffer containing 1 M NaCl and 1 M urea at 25°C and resuspended in 100 µL spiked in (100 pg)-containing RNA elution buffer (50 mM Tris–HCl pH 7.4, 5 mM EDTA, 10 mM DTT, 1% SDS). As a spike in for RNA purification control, a BDNF RNA from a mouse (kind gift of Dr. Sneha Shah, UMass Medical School) was used. Formaldehyde-induced crosslinks were reversed by incubation at 70°C for 30 min with mixing. The supernatant was mixed with TRIzol LS (Thermo Fisher) and coimmunoprecipitated RNA was purified according to the manufacturer's instructions. RT-PCR was performed as described above. Relative RNA enrichment was calculated as the ratio of normalized RNA levels in protein immunoprecipitation to levels in IgG immunoprecipitates.
Multiple sequence alignment
Fasta format sequences were retrieved from http://www.ncbi.nlm.nih.gov. Multiple sequence alignment was generated using T-Coffee (Notredame et al. 2000). The alignment was formatted using BoxShade v3.21.
Quantification and statistical analysis
Western blot signals were quantified in ImageJ. V1.53i. Luciferase assays, western blots, and polysome pellet results were measured in triplicate and analyzed in GraphPad Prism v9 for Windows. When two groups were compared, unpaired t-tests were applied, and when more groups were compared, a one-way ANOVA was applied. All data are presented as mean ± SD and replicate information is indicated in the figure legends (see Table 3).
Reagents and tools
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Shuying Sun (Johns Hopkins Medical School) for providing the C9ORF72 intronic G4C2 repeat reporter construct as well as the HeLa cell line expressing the construct, Dr. Gerhard Wagner (Harvard Medical School) for 4EGI, Dr. Sneha Shah (UMass Medical School) for the spike-in RNA control, Dr. Ruijia Wang and Sithara Raju Ponny (UMass Medical School) for help with bioinformatics, and Dr. Scott Shaffer of the UMass Proteomics Core for mass spectrometry. This work was supported by grants from TARGET ALS, the Muscular Dystrophy Association, and National Institutes of Health (NIH) R21NS109847 to J.D.R. and F.-B.G., NIH grants R01GM46779 and R01GM135087 to J.D.R., NIH grants R01CA207893 and R01CA248397 to R.J.S. and O.K., NIH grants R37NS057553 and R01NS101986 to F.-B.G., and NIH grants R21NS112766 and R21NS119952 to S.A.
Author contributions: J.D.R. and F.-B.G. conceived and supervised the project. E.S. performed the ribosome profiling and luciferase analysis. H.v.S. performed polysome analysis, DAP5 experiments, and luciferase analysis. S.A. cultured and differentiated the iPSC lines. O.K. and R.J.S. generated short hairpin vectors for knockdown. B.L. helped with bioinformatic analysis. K.S. performed 3D analysis of the uORF. H.v.S. and J.D.R. wrote the manuscript with contributions by all authors.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078963.121.
- Received August 24, 2021.
- Accepted October 31, 2021.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS



Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Heleen van ‘t Spijker, Sandra Almeida, and Emily Stackpole are the co-first authors of this paper, “Ribosome profiling reveals novel regulation of C9ORF72 GGGGCC repeat-containing RNA translation.” Heleen is a postdoctoral associate in Joel Richter's laboratory in the Department of Molecular Medicine, University of Massachusetts Chan Medical School. Her research focus is on the regulation of translation in neurological diseases, such as FTD/ALS and autism. Sandra is an Assistant Professor in the Department of Neurology at the University of Massachusetts Chan Medical School. Sandra's primary focus is elucidating the molecular mechanisms of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), using patient-derived human neurons to help uncover how FTD/ALS mutations impact cellular physiology and bring about their pathogenic consequences. Emily did this work as a postdoctoral associate in Joel Richter's laboratory, with a primary research focus on understanding the molecular mechanisms of neurological disorders, including C9ORF72-linked ALS/FTD.
What are the major results described in your paper and how do they impact this branch of the field?
We have investigated translation regulation of an expanded GGGGCC repeat in intron 1 of C9ORF72. Dipeptide repeat proteins accumulate in the brains of people carrying this intronic GGGGCC repeat expansion in the C9ORF72 gene. However, the mechanism by which RNAs containing these repeat expansions are translated to produce disease proteins is largely unknown. Our work, using ribosome profiling, identified translation start sites in the intron upstream of GGGGCC that can initiate translation independently of the repeats. This finding is in contrast with an earlier hypothesis proposing that translation is initiated by the direct association of the ribosomes with the G4C2 RNA. In addition, we found that protein synthesis takes place using unspliced RNA as the template and is regulated by both DAP5 and an upstream ORF that is conserved among primates. Altogether, these results expand our current understanding of the translational mechanisms involved in the production of disease proteins in C9ORF72-associated neurodegeneration.
What led you to study RNA or this aspect of RNA science?
HvS: I am a neuroscientist and I study plasticity in the brain. I became interested in investigating RNA to research how protein production is regulated in neurons. Control of translation in neurons makes it possible to produce proteins at the right time and in the right part of the neuron, for example at the synapse. Uncovering the pathways by which translation in neurons is regulated is key to improving our understanding of neuronal plasticity.
SA: With a focus on finding ways to prevent or block neurodegeneration in C9ORF72-associated FTD/ALS, an important step in that effort is to understand how the disease proteins are produced. Thus, elucidating the mechanisms involved in translation of the RNA harboring the repeats stood out as an important point in the pathogenic expression of the mutant protein and one that could potentially lead to new strategies to prevent the disease protein's synthesis at an early point.
ES: In studying the posttranscriptional mechanisms that underlie the production of mutant proteins in C9ORF72-linked ALS/FTD, our aim was to provide insight into the mechanistic underpinnings of the disease, which could ultimately help to identify therapeutic approaches toward curbing disease pathogenesis.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Our initial plan was to perform the majority of experiments in iPSC-derived neurons, but we encountered difficulties with low signal-to-noise and resolution. We therefore chose to pivot to using a reporter construct that provided much clearer and robustly interpretable results.
If you were able to give one piece of advice to your younger self, what would that be?
HvS: My advice for a younger self would be to ask for help more often and ask more different people for help. Science can feel isolating and it really helps to reach out to receive input from many points of view.
SA: I would advise the younger me to spend more time thinking about the big picture and the context of her experiments; selecting only the most relevant questions to go after before heading to the laboratory. In doing this, I'd tell her to seek advice and input from colleagues and peers outside of her field as their perspectives will almost inevitably sharpen and improve her own.
ES: I would encourage my younger self to be more intuitive—sometimes it's best to avoid over-rationalizing and instead simply follow the gut!
What are your subsequent near- or long-term career plans?
HvS: It is my ambition to become an independent neuroscientist and run a laboratory of my own, investigating neural plasticity and its role in neurological disorders.
What were the strongest aspects of your collaboration as co-first authors?
The strongest aspect of our collaboration was our disparate but complementing scientific backgrounds that empowered us to creatively and synergistically approach the same scientific question. Our lively discussions and shared perspectives on our data and the genesis of new experimental ideas were tremendously fun and greatly motivating.
How did you decide to work together as co-first authors?
HvS: The project was initiated by Dr. Stackpole and Dr. Almeida and I joined the project when I joined the Richter laboratory. Dr. Stackpole asked me to work with her on the project and I am very grateful she did, as it has been a great learning experience!








