
Exploring the epitranscriptome by native RNA sequencing
- 1Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, Barcelona 08003, Spain
- 2School of Biotechnology and Biomolecular Sciences, UNSW Sydney, Sydney, New South Wales 2052, Australia
- 3Universitat Pompeu Fabra, Barcelona 08002, Spain
- Corresponding authors: j.mattick{at}unsw.edu.au, eva.novoa{at}crg.eu
Abstract
Chemical RNA modifications, collectively referred to as the “epitranscriptome,” are essential players in fine-tuning gene expression. Our ability to analyze RNA modifications has improved rapidly in recent years, largely due to the advent of high-throughput sequencing methodologies, which typically consist of coupling modification-specific reagents, such as antibodies or enzymes, to next-generation sequencing. Recently, it also became possible to map RNA modifications directly by sequencing native RNAs using nanopore technologies, which has been applied for the detection of a number of RNA modifications, such as N6-methyladenosine (m6A), pseudouridine (Ψ), and inosine (I). However, the signal modulations caused by most RNA modifications are yet to be determined. A global effort is needed to determine the signatures of the full range of RNA modifications to avoid the technical biases that have so far limited our understanding of the epitranscriptome.
Keywords
RNA is chemically altered after transcription in more than 170 ways (Dal Magro et al. 2018), in all branches of life (Lorenz et al. 2017), and in the majority of RNA biotypes (Lim et al. 2015; Shoshan et al. 2015; Martínez-Pérez et al. 2017; Safra et al. 2017; Warda et al. 2017; Zhou et al. 2018; Begik et al. 2021). These chemical alterations, collectively termed the epitranscriptome (Saletore et al. 2012), cause changes in the shape and sometimes charge of the modified nucleotide, thereby affecting RNA structure, base-pairing and interactions with RNA-binding proteins (RBPs) (Harcourt et al. 2017). RNA molecules undergo modifications at different stages of differentiation and development, and in response to environmental and contextual cues, thereby altering splicing, export, regulatory functions, translation, and degradation (Zhao et al. 2014; Meyer et al. 2015; Du et al. 2016; Haussmann et al. 2016; Jonkhout et al. 2017; Warda et al. 2017; Kadumuri and Janga 2018; Delaunay and Frye 2019).
The addition and removal of RNA modifications are catalyzed by “writer” and “eraser” proteins, respectively (Shi et al. 2019; Begik et al. 2020; Esteve-Puig et al. 2020). Modified RNAs interact with “reader” proteins that recognize RNA modifications and trigger certain outcomes (Du et al. 2016; Haussmann et al. 2016; Yang et al. 2017; Shi et al. 2019). These proteins are collectively known as RNA-modifying proteins (RMPs), the dysregulation of which can have drastic consequences such as cancer, infertility, obesity, and neurological diseases (Torres et al. 2014; Jonkhout et al. 2017; Kadumuri and Janga 2018; Delaunay and Frye 2019; Esteve-Puig et al. 2020). The repertoire of RNA modification types and substrate range (which includes mRNAs, tRNAs, rRNAs, and many other noncoding RNAs) has been expanded by successive gene duplications of RMPs that have occurred at the base of the eukaryote, metazoan, vertebrate, and primate lineages, with more than 90 RMP writers currently cataloged in the human genome (Begik et al. 2020; Boccaletto et al. 2022).
Before the advent of next-generation sequencing (NGS), our knowledge of the epitranscriptome came mainly from transfer RNA (tRNA) and ribosomal RNA (rRNA). The abundance of these RNA molecules, as well as their high modification stoichiometries, made it possible to map and characterize their RNA modifications using low-throughput methods such as thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC) (Grosjean et al. 2007; Köhrer and Rajbhandary 2008; Jora et al. 2019), and liquid chromatography-tandem mass spectrometry (LC–MS/MS) (Helm and Motorin 2017; Jora et al. 2019).
The most studied RNA modifications, directly or indirectly, are N6-methyladenosine (m6A), 5-methylcytosine (m5C), 2′-O-methylation (Nm), pseudouridine (Ψ), and the deaminated version of adenosine, inosine (I). Disruptions of m6A pathways have been associated with defects in embryo development, differentiation, neural function, sex determination, spermatogenesis, adipogenesis, and cancer in many organisms including human, mouse, zebrafish, and flies (Zheng et al. 2013; Batista et al. 2014; Wang et al. 2014; Zhao et al. 2014; Geula et al. 2015; Zhang et al. 2015; Haussmann et al. 2016; Lence et al. 2016; Zhao et al. 2017). Furthermore, targeting the m6A pathway with small molecule inhibitors can lead to a reduction in cancer progression (Roundtree et al. 2017; Yankova et al. 2021).
Most of our understanding of the roles of m6A and m5C comes from loss-of-function studies of the RNA methyltransferases involved. In mammals, there are multiple m6A writers (typically METTL3 for mRNAs [Liu et al. 2014; Haussmann et al. 2016], METTL5 for rRNAs [van Tran et al. 2019] and METTL16 for snRNAs [Warda et al. 2017]), as well as readers and erasers (Meyer and Jaffrey 2017). METTL3 is responsible for cotranscriptionally methylating mRNAs with m6A (Schwartz et al. 2014b), leading to altered stability of the mRNAs (Dierks et al. 2021). METTL3 also regulates heterochromatin in embryonic stem cells (Xu et al. 2021) and promotes homologous recombination-mediated repair of double-strand breaks by modulating DNA–RNA hybrid accumulation (Zhang et al. 2020). METTL16, which is essential for mouse embryonic development (Mendel et al. 2018), regulates the expression by intron retention of an enzyme that produces the methyl donor, S-adenosyl methionine (Pendleton et al. 2017). METTL5-mediated m6A addition to 18S rRNA is essential for the translational machinery (Ignatova et al. 2020), and its absence is associated with neural function defects (Wang et al. 2022) and cardiac hypertrophy (Han et al. 2022). On the other hand, the m6A reader YTHDC1 interacts with LINE1 RNA and alters its scaffolding function in mouse ESCs and early embryos (Chen et al. 2021), while PRRC2A, another m6A reader, is involved in oligodendroglial specification and myelination (Wu et al. 2019). The ALKBH5 m6A eraser controls translation (Liu et al. 2016) and the splicing and stability of long 3′UTR mRNAs in male germ cells (Tang et al. 2018). On the other hand, the FTO demethylase, which has been reported to act both on m6A and m6Am (Jia et al. 2011; Mauer et al. 2019), is required for proper mRNA splicing (Zhao et al. 2014; Merkestein et al. 2015) and snRNA processing (Mauer et al. 2019).
There are eight mammalian m5C writers, NSUN1-7 and DNMT2. NSUN1, 2, 5 and DNMT2 are present in all eukaryotes, whereas other NSUN members are specific to multicellular organisms and are differentially expressed during development (Chi and Delgado-Olguín 2013), particularly in the brain: mutations in the NSUN2 gene have been associated with autosomal-recessive intellectual disability (Khan et al. 2012), Dubowitz-like syndrome (Martinez et al. 2012), disrupted neurogenesis and impaired brain development (Flores et al. 2017). Mutations in NSUN3 have been related to mitochondrial deficiency, leading to developmental disorders in humans (Van Haute et al. 2016) and impaired differentiation of embryonic stem cells in mice (Trixl et al. 2018). Finally, NSUN7, which is predominantly expressed in testis, has been shown to play an important role in male fertility, although the molecular mechanism underlying this role is yet to be determined (Harris et al. 2007). Recent studies have disclosed the target substrates for some of the members of this family: NSUN1/NOP2 and NSUN5 target rRNAs (Schosserer et al. 2015; Heissenberger et al. 2020), NSUN2 targets tRNAs and vault RNA (Sajini et al. 2019), NSUN3 methylates mitochondrial tRNAs (Van Haute et al. 2016) and NSUN6 methylates mRNAs (Selmi et al. 2021).
The effect of Ψ modification on mRNA stability is controversial (Schwartz et al. 2014a; Nakamoto et al. 2017), but it is established that the presence of Ψ modifications in synthetic mRNAs increases their evasion of the innate immune system and enhances their translation, the basis of the new generation of mRNA vaccines (Karikó et al. 2005, 2008, 2012; Warren et al. 2010), including COVID-19 vaccines that take advantage of N1-methylated pseudouridine (m1Ψ) for increased efficiency (Andries et al. 2015; Nance and Meier 2021). Moreover, studies on pseudouridine synthases have shown their association with multiple human autoimmune diseases including celiac disease (Festen et al. 2011), X-linked ichthyosis (Preumont et al. 2010), Crohn's disease (Festen et al. 2011), and mitochondrial myopathy and sideroblastic anemia (Patton et al. 2005).
Being a common modification, 2′-O-methylations (Nm) are involved in many biological processes. Guided by small nucleolar RNAs (snoRNAs) that recognize specific sequences, Nm modifications are placed by RMPs such as fibrillarin (FBL) or members of the FTSJ family (Dimitrova et al. 2019). Loss-of-function of the 2′-O-methyltransferase FBL enzyme results in disruption of translation (Erales et al. 2017), whereas its overexpression leads to enhanced translation and increased proliferation of breast cancer cells (Marcel et al. 2013). 2′-O-methylation is also important for spliceosome assembly and function due to its presence in snRNAs (Dönmez et al. 2004; Karunatilaka and Rueda 2014), and its absence in U6 snRNA leads to alterations in splicing and impaired spermatogenesis in mice (Wang et al. 2020).
An abundant modification in the mammalian brain, especially human, is the deamination of adenosine to inosine (A-to-I), also termed RNA “editing,” which is catalyzed by adenosine deaminases acting on RNA (ADARs) (Paz-Yaacov et al. 2010). RNA editing is known to occur in a number of neuroreceptor mRNAs, but also thousands of other RNAs (Tan et al. 2017), likely playing a major role in neuronal plasticity (Mattick and Mehler 2008). ADARs can shuttle between the nucleus and the cytoplasm upon RNA binding (Fritz et al. 2009), and have been shown to accumulate in the nucleus during neuronal development (Behm et al. 2017). Moreover, inosine acts as a mark for self-RNA (Mannion et al. 2014; Liddicoat et al. 2015), preventing aberrant activation of antiviral RIG-I-like receptors (Quin et al. 2021), holding great promise for future RNA-based therapeutics (Sahin et al. 2014). In addition to ADAR proteins, additional RMPs catalyze A-to-I deamination in mammalian cells: (i) ADAD1 and ADAD2, which are testis-specific and whose targets are unknown, and (ii) adenosine deaminases that act on tRNAs (ADATs), which are evolutionarily conserved in both prokaryotes and eukaryotes (Schumacher et al. 1995; Hough and Bass 1997; Gerber and Keller 2001) and play a key role in translation by expanding the wobbling capacity of the modified tRNA anticodons.
The field is in its infancy. To analyze the epitranscriptome globally, high-throughput methodologies are required. Until recently, the mapping of modifications in RNA was accomplished mainly by using modification-specific antibodies to immunoprecipitate and enrich modified sites before sequencing (Dominissini et al. 2012, 2016; Meyer et al. 2012; Safra et al. 2017). Methylated RNA immunoprecipitation followed by sequencing (MeRIP-seq) was initially used for m6A modification and showed an unexpected abundance and dynamic regulation of m6A modifications in mRNAs (Dominissini et al. 2012; Meyer et al. 2012).
NGS relies on the sequencing by synthesis method, which involves reverse transcription (RT) of RNA into cDNA, followed by a second strand synthesis with fluorophore tagged DNA molecules (Slatko et al. 2018). Therefore, the presence of RNA modifications cannot be detected directly with NGS techniques. Some modifications (m1A, m3C, m3U, m1G, m2,2G, m1acp3Y), however, cause disruption in Watson–Crick base pairing, leading to reverse transcription errors such as RT drop-off or increased error frequencies (deletions, insertions and mismatches) (Motorin and Helm 2019; Mongan et al. 2019) at the modified sites, which has proven useful for the detection of these modifications (Motorin et al. 2007; Ryvkin et al. 2013; Hauenschild et al. 2015; Zheng et al. 2015; Tserovski et al. 2016; Potapov et al. 2018). Alternatively, chemical treatment of RNA with reagents that selectively react with modified bases will stall reverse transcription (Moshitch-Moshkovitz et al. 2022), and the analysis of read ends can be used to locate the RNA modifications. Examples of such protocols include CMCT treatment for Ψ (Carlile et al. 2014; Lovejoy et al. 2014; Schwartz et al. 2014a), bisulfite treatment for m5C (David et al. 2017), sodium cyanoborohydride treatment for ac4C (Thalalla Gamage et al. 2021), hydrazine and/or aniline treatment for m3C (Marchand et al. 2018; Cui et al. 2021) and glyoxal treatment for inosine (Cattenoz et al. 2013).
Despite their significant contribution to the field, NGS-based approaches have a number of limitations. Limited availability of specific chemical reagents or antibodies restricts the number of RNA modifications that can be detected (Motorin and Helm 2019; Anreiter et al. 2020). Moreover, even when an antibody is available for a specific modification, cross-reactivity with other modifications is a serious problem exposed by studies reporting a high amount of false positives (Schwartz 2018; Grozhik et al. 2019). There are also other limitations due to the nature of NGS techniques, which involve reverse transcription and PCR amplification. Moreover, these techniques yield only relatively short reads, which typically eliminates the possibility of detecting modifications that are isoform-specific or co-occurring in the same transcript (Schaefer et al. 2017). PCR amplification and multiple ligation steps also introduce biases in quantifying modifications at specific sites (Lahens et al. 2014; Schaefer et al. 2017).
Over a decade ago, Pacific Biosciences (PacBio) introduced a new “third generation” sequencing (TGS) platform based on kinetic changes of reverse transcription detected by zero-mode waveguide (ZMW) technology (Check Hayden 2009). Eventually, the ZMW technology was used to detect m6A modifications in full-length RNA sequences (Vilfan et al. 2013; Potapov et al. 2018), but no further development has been reported.
Nanopore sequencing technologies, such as those developed by Oxford Nanopore Technologies (ONT), have the potential to provide a more generalizable solution (Novoa et al. 2017; Garalde et al. 2018). Nanopore sequencing relies on the measurement of alterations in ionic current when a nucleic acid transits a protein nanopore embedded in the membrane of a flowcell (Fig. 1A; Jain et al. 2016; Lu et al. 2016). This long-read TGS technology can directly sequence native RNAs, and, in principle, allows the detection of multiple RNA modifications at the single-nucleotide and single-molecule resolution (Garalde et al. 2018), provided there is an altered signal that can be distinguished from the unmodified nucleotide (Fig. 1B).
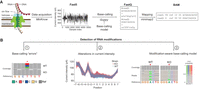
An overview of using direct RNA sequencing to detect RNA modifications. (A) After performing RNA library preparation (ligation of the RNA to a helicase-containing DNA adapter, plus optional reverse transcription step), RNA molecules can translocate through the nanopore embedded on a membrane with the help of a helicase protein, at an approximate speed of 70 bases per second (under the library preparation kits: SQK-RNA001 and SQK-RNA002). RNA translocation disrupts the current created by the ion flow that is passing through the nanopore. The current intensity information is acquired and processed using MinKnow software, which generates Fast5 files containing the current intensity information of each identified read (one Fast5 file will contain up to 4000 reads). Fast5 files can then be base-called using base-calling algorithms (e.g., Guppy, Bonito), which are fed with a base-calling model, generating FastQ files as output. FastQ files can be mapped to a reference sequence by using an alignment tool (e.g., minimap2), which creates a BAM file. Modification information can be stored in BAM files using specific tags. (B) Schematic overview of three major strategies that can be used to detect RNA modifications in direct RNA nanopore sequencing data. A first strategy consists in identifying RNA modifications in the form of nonrandom base-calling “errors,” which can be seen in the form of increased base-calling “errors” (mismatch, insertion and deletion) at the modified site (position 0) and/or surrounding positions (left panel). In this strategy, the use of knockout and/or knockout strains allows distinguishing “errors” caused by the presence of RNA modifications from those that are intrinsic to the sequencing and or base-calling itself (i.e., background “error”). A second strategy involves using raw current intensity (signal intensity, dwell time and/or trace features) to identify positions with altered current intensity values, when comparing two strains (e.g., wild type and knockout) or when comparing reads within a given sample (middle panel). A third strategy consists in using a modification-aware base-calling model (instead of the canonical model that predicts four letters) when performing the base-calling step (right panel). This approach requires generating training sets that can be used to train the modification-aware base-calling model.
A pioneering study using direct ONT RNA sequencing showed that m6A sites exhibit different current signals than unmodified sites in the same position in synthetic RNAs (Garalde et al. 2018). Shortly afterward, direct RNA sequencing of bacterial 16S rRNA confirmed that changes in current intensity were also observed at m7G-modified sites (Smith et al. 2019). Later studies showed that alterations in current intensity are also reflected in the base-calling data in the form of mismatches, insertions, and deletions (Liu et al. 2019). These findings triggered several subsequent studies to detect m6A and Ψ modifications using base-calling “error signatures” from nanopore sequencing data (Fig. 1B, right panel; Liu et al. 2019; Viehweger et al. 2019; Parker et al. 2020; Begik et al. 2021; Jenjaroenpun et al. 2021; Abebe et al. 2022). Furthermore, several studies took advantage of the current signal metrics to detect modified sites by comparison with a paired sample with fewer or no modifications (Fig. 1B, middle panel; Begik et al. 2021; Leger et al. 2021), using internal unmodified sequences from the same sequencing run (Ramasamy et al. 2022) or implementing machine learning approaches to determine the proportion of modified molecules from single samples (Begik et al. 2021; Pratanwanich et al. 2021).
Information obtained from full-length native RNA nanopore sequencing has been used to detect isoform-specific modifications (Aw et al. 2021), as well as to predict RNA modifications in individual reads (Begik et al. 2021; Acera Mateos et al. 2022), providing a resolution of the epitranscriptome at an unprecedented level, which had not been obtained using short-read sequencing data (Leger et al. 2021). With the help of computational tools, unmodified and modified reads can be distinctly classified based on their unique current signal signatures, making it possible to detect even slight changes in the proportion of modified molecules, referred to as modification stoichiometry, across different conditions and cell types (Begik et al. 2021; Pratanwanich et al. 2021). Moreover, a recent method termed nano-COP demonstrated that nascent RNAs could be sequenced using direct RNA nanopore sequencing, providing a promising framework that could be used in the future to further investigate the interplay between splicing and cotranscriptional RNA modifications (Drexler et al. 2020).
The opportunity to expand the repertoire of epitranscriptomic modifications that can be studied using direct RNA nanopore sequencing, however, is limited by the lack of available data by which to recognize the signals of different RNA modifications in nanopore sequencing signals. This recently led to a call to action to identify all modifications in full-length RNAs (Alfonzo et al. 2021). This is not a straightforward task, as the signal typically reflects the transit of five nucleotides that transit the pore at any one time, so complex signal processing and analysis software, often requiring machine learning algorithms, is required (Lorenz et al. 2020; Begik et al. 2021; Furlan et al. 2021; Leger et al. 2021; Liu et al. 2021; Pratanwanich et al. 2021; Wan et al. 2022). A general solution, in principle, is to construct training sets for nanopore sequencing using synthetic sequences that contain all possible 5-mers that are transcribed in vitro with either unmodified (A, C, G, U) or modified nucleotides. Indeed, this approach was the basis for the “curlcakes,” synthetic sequences covering all possible 5-mers with minimized RNA secondary structure (Liu et al. 2019). These and similar data sets were used to identify the base-calling signatures of m6A, I, m5C, Ψ, m1G, m2,6A, m7G, and Nm modifications, which proved valuable to predict RNA modifications de novo (Begik et al. 2021; Jenjaroenpun et al. 2021; Nguyen et al. 2022). However, the generation of such sequences for the whole repertoire of RNA modifications is not a simple matter, as most RNA modifications cannot be synthesized chemically or enzymatically, limiting the set of RNA modifications for which we can train machine learning algorithms.
Even when these sequences can be generated, they still pose significant challenges. Firstly, training models on highly modified (100% stoichiometry) data sets, which are the simplest to build and for which the “ground truth” is known, will leave a substantial amount of k-mers uncovered. For example, the 5-mer AGm6ACA, which is a naturally occurring m6A modification sequence context, will actually be m6AGm6ACm6A in the 100% modified training set; consequently, models trained with 100% modified sequences will only be applicable to a subset of biologically relevant sequence contexts. A solution to avoid multiple modifications in the same k-mer would be to construct training sets that are in vitro transcribed with low concentration of modified nucleotides, relative to the corresponding unmodified counterpart. However, these data sets would require knowing which site is modified or unmodified in each molecule, to then train the training set adequately. It is also likely that the k-mer length used by the machine learning algorithm is longer than the 5-mer region that is located in the nanopore. In this regard, several studies have shown that the dwell time of bases upstream to the modified site (e.g., positions −10 to −12) can be altered, in addition to the signal intensity at the 5-mer region (Begik et al. 2021; Stephenson et al. 2022). These limitations pose important challenges to generate models that can predict RNA modifications de novo.
To date, all methods developed to detect RNA modifications, including those relying on base-calling “errors” and/or signal intensity differences, require that base-calling, mapping and/or signal event “resquiggling” are performed first, and then, the RNA modifications can be predicted (Stoiber et al. 2017; Liu et al. 2019; Lorenz et al. 2020; Parker et al. 2020; Price et al. 2020; Begik et al. 2021; Jenjaroenpun et al. 2021; Leger et al. 2021; Pratanwanich et al. 2021; Hassan et al. 2022). However, these approaches typically suffer both from stoichiometry biases (lowly modified sites are more poorly predicted) and sequence coverage biases (minimum coverage of ∼30–50 reads is required to split the populations into “modified” and “unmodified”). An alternative approach that could overcome these limitations, and detect RNA modifications in individual RNA reads and in a stoichiometry- and coverage-independent manner, would consist of developing novel RNA base-calling models that would directly predict native RNA modifications from the nanopore signal features (Fig. 1B, right panel). Software such as taiyaki (https://github.com/nanoporetech/taiyaki) or flappie (https://github.com/nanoporetech/flappie) could be used to generate modification-aware RNA base-calling models, which would be used instead of the canonical base-calling model. To this end, adequate training data sets are also required, with and without the RNA modifications of interest, the generation of which still remains a major challenge that must be solved in the years to come, as explained above.
The generation of novel base-calling RNA models constitutes a promising solution for the detection of RNA modifications in the near future, as it solves both the stoichiometry and coverage limitations that current methods suffer from. RNA base-calling models, however, are unlikely to be successful for simultaneous detection of >170 different RNA modifications under the same base-calling model, as certain RNA modifications may not produce alterations in the current intensity that are sufficiently distinct from all other RNA modifications. To overcome this limitation, recent efforts have transitioned toward separating the process of base-calling from the process of modification detection. Indeed, in this approach, RNA modifications are predicted in a two-step process: In the first step, RNA would be base-called under a canonical RNA base-calling model (modification-unaware), and in the second step, each nucleoside would be predicted as modified or unmodified, using toolkits such as remora (https://github.com/nanoporetech/remora). Models to predict RNA modifications using this latter approach, however, have so far not been released. Whether this approach will be more successful at predicting and distinguishing over 170 different RNA modifications that are described to date is yet to be determined.
Developing modification-aware base-calling models (or alternative approaches) for all modifications is a huge undertaking, but feasible if there was a global effort to do so, such as the Human Epigenome Consortium (Stunnenberg et al. 2016), which would change the study of the epitranscriptome from glimpses to comprehensive analysis, something that has eluded the field of epigenetics. Nanopore sequencing makes this objective achievable, without which our understanding of the epitranscriptome will be constrained. An important objective in RNA biology is to map all RNA modifications in cell biology, development, environmental adaptation and brain function, which constitutes a fundamental challenge that could and should be addressed in a collaborative manner. In this regard, a great opportunity to establish a collaborative effort of this caliber will be the upcoming RNA Society meeting, which will be held in Singapore in 2023.
ACKNOWLEDGMENTS
O.B. is supported by a MERCK 2020 Research Drug Discovery Grant. We thank Sonia Cruciani and Leszek Pryszcz for providing some figure panels used to build Figure 1. We thank all the Novoa laboratory for their helpful discussions and brainstorming sessions on approaches to detect RNA modifications using nanopore sequencing. This work was supported by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) (PID2021-128193NB-100 to E.M.N.), the European Research Council (ERC-2021-STG no. 101042103 to E.M.N.), the AECC Scientific Foundation (LABAE211658NOVO to E.M.N.), and UNSW Sydney (SHARP Professorial Fellowship to J.S.M.). We acknowledge the support of the MEIC to the EMBL partnership, Centro de Excelencia Severo Ochoa and CERCA Programme/Generalitat de Catalunya.
Footnotes
-
Editor's Note: Oğuzhan Beğik was the recipient of the 2022 Eclipse Award for Innovation in High Throughput Biology presented by the RNA Society.
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079404.122.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available undera Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








