
Determination of ribonuclease sequence-specificity using Pentaprobes and mass spectrometry
- Joanna L. McKenzie1,
- Johanna M. Duyvestyn1,
- Tony Smith2,
- Katerina Bendak3,
- Joel MacKay3,
- Ray Cursons1,
- Gregory M. Cook4 and
- Vickery L. Arcus1,5
- 1Department of Biological Sciences,
- 2Department of Computer Science, University of Waikato, Hamilton 3240, New Zealand
- 3School of Molecular and Microbial Biosciences, University of Sydney, NSW 2006, Australia
- 4Department of Microbiology and Immunology, Otago School of Medical Sciences, University of Otago, Dunedin 9054, New Zealand
Abstract
The VapBC toxin-antitoxin (TA) family is the largest of nine identified TA families. The toxin, VapC, is a metal-dependent ribonuclease that is inhibited by its cognate antitoxin, VapB. Although the VapBCs are the largest TA family, little is known about their biological roles. Here we describe a new general method for the overexpression and purification of toxic VapC proteins and subsequent determination of their RNase sequence-specificity. Functional VapC was isolated by expression of the nontoxic VapBC complex, followed by removal of the labile antitoxin (VapB) using limited trypsin digestion. We have then developed a sensitive and robust method for determining VapC ribonuclease sequence-specificity. This technique employs the use of Pentaprobes as substrates for VapC. These are RNA sequences encoding every combination of five bases. We combine the RNase reaction with MALDI-TOF MS to detect and analyze the cleavage products and thus determine the RNA cut sites. Successful MALDI-TOF MS analysis of RNA fragments is acutely dependent on sample preparation methods. The sequence-specificity of four VapC proteins from two different organisms (VapCPAE0151 and VapCPAE2754 from Pyrobaculum aerophilum, and VapCRv0065 and VapCRv0617 from Mycobacterium tuberculosis) was successfully determined using the described strategy. This rapid and sensitive method can be applied to determine the sequence-specificity of VapC ribonucleases along with other RNA interferases (such as MazF) from a range of organisms.
Keywords
INTRODUCTION
The Pfam database lists 4089 proteins belonging to the PIN-domain family (PF01850) from 1129 different species (Finn et al. 2010), including eukaryotes, archaea, and prokaryotes. The VapBC (virulence-associated protein) toxin-antitoxin (TA) family are defined by their toxic components, which belong to the PIN-domain protein family. TA pairs are arranged as a bicistronic operon (Anantharaman and Aravind 2003), and the toxin gene encodes a stable toxin (a VapC PIN-domain). The preceding gene encodes a labile antitoxin (VapB) that binds to and inhibits the activity of the toxin. In general, the antitoxin also binds to DNA via its N-terminal domain and interacts with specific sequences within the promoter of the TA operon (Gerdes et al. 1986). The term “RNA interferase” has been given to the VapC toxins, reflecting their biochemical activity as sequence-specific RNases. This term also encompasses an unrelated TA toxin family, the MazF proteins, that cleaves mRNA (Zhang et al. 2003).
The VapBC TAs are the largest family of the nine TA families classified (Gerdes et al. 2005; Van Melderen et al. 2009), yet they are the least well characterized. Although sequence conservation across PIN-domains is poor, there is conservation of the three-dimensional structure, which results in a clustering of acidic residues proposed to constitute the active site (Arcus et al. 2011). Of the few VapC/PIN-domain proteins characterized, most demonstrate ribonuclease activity (Daines et al. 2007; Miallau et al. 2009; Ramage et al. 2009; Ahidjo et al. 2011). VapC of Shigella flexneri 2a virulence plasmid pMYSH6000 and VapC of Salmonella enterica serovar Typhimurium T2 are site-specific tRNases that cleave initiator tRNA between the anticodon stem and loop (Winther and Gerdes 2011). VapC6 from Sulfolobus solfataricus targets mRNAs involved in the heat shock response (Maezato et al. 2011), and VapC from nontypeable Haemophilus influenzae degrades free RNA in vitro but not double-stranded (ds) DNA or single-stranded (ss) DNA (Daines et al. 2007). VapC-5 from Mycobacterium tuberculosis displays Mg2+-dependent ribonuclease activity (Miallau et al. 2009), although the catalytic mechanism or sequence-specificity of these enzymes has not been determined. Although FitB from Neisseria gonorrhoeae structurally resembles VapCPAE2754, its ribonuclease activity has not been characterized due to an inability to express soluble recombinant protein (Mattison et al. 2006). It has, however, been shown to mediate intracellular trafficking in vivo (Hopper et al. 2000). PIN-domain proteins from eukaryotes are associated with nonsense mediated decay (NMD) of RNA (Huntzinger et al. 2008) and processing of pre-18S rRNA fragments (Lamanna and Karbstein 2009).
An important step to understanding the function of VapC and the PIN-domain proteins is the biochemical elucidation of their RNase sequence-specificity. We have previously demonstrated ribonuclease activity for VapCRv0065 and VapCRv0617 from M. tuberculosis (Ahidjo et al. 2011). Here, we describe a detailed protocol for the purification of VapC from two organisms and the determination of VapC sequence-specificity as a first step in identifying the biological substrate for VapC enzymes (Fig. 1). There are two significant hurdles in the biochemical characterization of VapC: expression and purification of VapC (in many cases, a very toxic protein) and determination of the target RNA sequence. To overcome the first hurdle, we have developed an approach to overexpress and purify the inactive VapBC complex and then isolate VapC alone, from this complex. This circumvents the requirement to express the toxic VapC alone, which has previously been shown to be problematic (Mattison et al. 2006; Miallau et al. 2009; Ramage et al. 2009). To overcome the second hurdle, we have developed a ribonuclease assay using synthetic substrates and MALDI-TOF mass spectroscopy (MS).

Schematic diagram of VapC purification, sequence-specificity, and kinetic analysis. To neutralize VapC toxicity it is expressed in complex with VapB. Proteolyticly sensitive VapB is removed by trypsin, and VapC is purified by anion exchange chromatography. RNase activity and sequence-specificity are screened using Pentaprobes (ticks indicate Pentaprobes cut by all VapC proteins tested; crosses indicate Pentaprobes not cut). Short RNA oligonucleotides are designed based on the Pentaprobe RNA cleaved by all VapC proteins and cut sites determined by MALDI-TOF MS and in-house software. A fluorogenic RNA substrate with a single cut site is then designed for determination of VapC kinetic parameters.
Although determining general ribonuclease activity is straightforward, there are few methods for elucidation of sequence-specificity. The most common method used is primer extension (Zhang et al. 2003; Zhu et al. 2009; Han et al. 2010). Alternatively, RNA transcripts of specific genes can be assayed and cleavage sites determined by labeling the assay products with a radioactive isotope such as 32P (Munoz-Gomez et al. 2004; Fu et al. 2007). However, these approaches require prior knowledge of a bona fide target mRNA and/or use the viral MS2 RNA as a substrate. We have developed a new rapid ribonuclease activity assay for sensitive determination of the sequence-specificity of VapC proteins. Identification of VapC target sequences allows determination of possible in vivo substrates for VapC. This new approach employs the use of Pentaprobes instead of the commonly used MS2 bacteriophage RNA (Zhu et al. 2006; Ramage et al. 2009). Although MS2 RNA is complex enough to determine cleavage sites longer than three bases (i.e., MS2 RNA is 3569 bases in length and contains an approximately equal number of each base), it forms an extensive secondary structure, requiring the use of CspA (a major cold shock protein from Escherichia coli) as an RNA chaperone (Jiang et al. 1997). The Pentaprobes were generated using a computer algorithm that determined the minimum sequence required to cover every combination of five bases—516 base pairs (Kwan et al. 2003). This sequence was then encoded in six overlapping dsDNA molecules from which RNA can be transcribed, resulting in 12 ssRNA segments (six in the forward direction and six complementary sequences in the reverse direction) covering every combination of five bases (Bendak et al. 2012). The reverse Pentaprobe sequences also allow dsRNA to be synthesized. The use of Pentaprobe RNA, which is shorter than the commonly used MS2 RNA, reduces the amount of secondary structure present in the RNA substrate (negating the need for an RNA chaperone) while still allowing screening of recognition sequences up to five bases. Other advantages of the Pentaprobe plasmids are that dsDNA, ssDNA, dsRNA, and ssRNA can be synthesized and that biochemical activity is tested against all substrate types.
Although there have been reports of MALDI-TOF MS being used for analysis of RNA up to 461 nucleotides (nt) (Kirpekar et al. 1994), mass resolution decreases dramatically with increasing oligonucleotide size. Therefore to determine the RNase sequence-specificity, we designed short overlapping RNA oligonucleotides based on the targeted Pentaprobe sequence (Fig. 1). These short RNAs were cut and analyzed using MALDI-TOF MS. The use of RNA oligonucleotides and MALDI-TOF MS negates the need for a reverse transcriptase. MS provides a fast, sensitive, and direct way of analyzing the products of ribonuclease activity assays.
Once the specificity of VapC was known, detailed kinetic analyses were carried out using a sensitive fluorometric substrate similar to that of the method described by Kelemen et al. (1999) and Wang and Hergenrother (2007) for quantitation of VapC activity and kinetic analyses of these enzymes.
This approach is fast, robust, and general and can be combined with results from biological assays to link the in vitro and in vivo activities of this large and important family of enzymes. It is also amenable to other sequence specific RNases such as the MazF family of RNases.
RESULTS AND DISCUSSION
Expression and purification of VapC
VapC proteins are generally toxic and difficult to overexpress. We surveyed a range of approaches to overexpress VapC. For example, initial attempts to express VapCMSMEG_1284 in its native host Mycobacterium smegmatis were unsuccessful due to its toxicity. Following induction, no VapC protein was detected on an SDS-PAGE gel, and sequencing of the VapC expression construct revealed a 2-bp insertion that was not present prior to transformation (resulting in a nonsense stop-codon downstream), explaining the absence of VapC protein expression in this case. This mutation presumably arises in response to basal expression of toxic VapC from the leaky T7 promoter before induction and a requirement to escape from this toxic expression in order for the bacteria to grow.
Expression of VapCMSMEG_1284 from M. smegmatis as a His-tagged fusion protein in E. coli gave insoluble protein. However, expression of VapCMSMEG_1284 as a maltose binding protein (MBP) fusion in E. coli greatly increased the solubility of VapC. This approach was reported by Ramage et al. (2009) for the expression of M. tuberculosis VapC proteins. When in the context of a fusion protein, MBP acts as a general molecular chaperone and promotes proper folding of the attached protein (Kapust and Waugh 1999). MBP most probably prevents dimerization of VapC, thereby abolishing its activity and its toxicity to the cell and enabling soluble protein expression. Although we found MBP-VapC in the soluble fraction of the E. coli lysate, the fusion protein elutes in the void of a size-exclusion column, indicating that it forms large soluble aggregates. Further, proteolytic cleavage of MBP from VapC was not possible, perhaps as a consequence of this aggregation (data not shown).
In contrast, the VapBC complex is inactive and thus more amenable to overexpression (Robson et al. 2009). It is a well-known feature of TA systems that the antitoxin is less stable and more susceptible to proteolytic degradation compared with the toxin (Gerdes et al. 2005). To exploit these different properties of VapB and VapC, the protease trypsin was used to digest away VapB from several recombinant VapBC complexes. Thus the mycobacterial VapC proteins VapCRv0065 and VapCRv0617 (from M. tuberculosis) were isolated by digestion of their cognate VapBC complexes with 0.1 mg/mL trypsin and subsequent anion exchange purification (Fig. 2). The protease reaction was stopped by the addition of 0.1 mg/mL trypsin inhibitor (from Soybean max) before purification. MALDI-TOF MS of the digestion mixture, following purification of VapC by anion exchange chromatography, showed two masses corresponding to VapC with and without the C-terminal His-tag (Fig. 2).
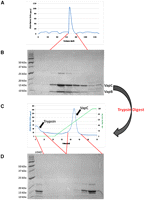
Purification and isolation of VapC. (A) The VapBC complex Rv0065a-Rv0065 from M. tuberculosis expressed in M. smegmatis elutes as a single peak on an S200 size exclusion column. (B) The corresponding SDS-PAGE gel shows the presence of both VapB and VapC proteins in the complex. (C) Trypsin digestion of the VapBC complex followed by anion exchange chromatography. Due to differences in their isoelectric points, trypsin is eluted early from the column while VapC is eluted later in the NaCl gradient. (D) Corresponding SDS-PAGE gel demonstrates the trypsin digest of VapBC loaded onto the column (LOAD) and the presence of VapC with and without the C-terminal His-tag.
VapCPAE2754 and VapCPAE0151 (from the crenarchaeon Pyrobaculum aerophilum) can be overexpressed in E. coli (Arcus et al. 2004; Bunker et al. 2008) with no toxicity problems as seen for other VapC proteins. This is most probably a result of their very low solubility at neutral pH. Using a lysis buffer at pH 9.2 resolubilizes VapC from the cell lysate.
Screening for VapC sequence-specificity using Pentaprobes
The 12 Pentaprobe vectors can be used to make ss and ds RNA and DNA substrates that encode every combination of five bases (Bendak et al. 2012). We have demonstrated ribonuclease activity for VapCRv0065 and VapCRv0617 using one of the ss Pentaprobe RNA substrates (Ahidjo et al. 2011). Here we describe a detailed protocol for determination of the sequence-specificity of RNases and use this approach to determine the sequence-specificity of two VapCs from M. tuberculosis and two from P. aerophilum. Figure 3 illustrates the use of Pentaprobe RNAs for screening of VapC sequence-specificity. VapCPAE0151 and VapCPAE2754 are sequence-specific and target the same sequence as shown by the same cleavage pattern for the 924 Pentaprobe RNA (Fig. 3). VapCRv0065 and VapCRv0617 target a different sequence on the same 924 Pentaprobe substrate (Fig. 3). The sequence-specificity of VapCPAE0151, VapCPAE2754, VapCRv0065, and VapCRv0617 also differ from that of VapCMSMEG_1284, which does not cut the 924 Pentaprobe RNA (the sequence-specificity and biological role of VapCMSMEG_1284 are published elsewhere) (McKenzie et al. 2012). VapB in complex with VapC inhibits its RNase activity as shown in Figure 3 (the VapBC complex PAE2754/PAE2755 could not be tested due to the insolubility of VapB upon expression).
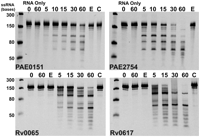
VapC proteins Rv0065, Rv0617, PAE0151, PAE2754 display sequence-specific, Mg2+-dependent ribonuclease activity. VapCPAE0151 and VapCPAE2754 appear to target the same sequence as seen by the same RNA fragments on the gels; this is also seen for VapCRv0065 and VapCRv0617, which also appear to target the same sequence. Negative controls (RNA only at 0- and 60-min time points) show no ribonuclease contamination; 12 mM EDTA (E) inhibits VapC activity, and VapC when in complex with VapB (C) displays no ribonuclease activity. This lane is missing from the VapCPAE2754 panel as VapBC cannot be produced in this case due to the insolubility of VapB. Ladder indicates sizes of RNA.
VapCPAE0151 and VapCPAE2754 exhibit no nuclease activity against dsRNA, ssDNA, and dsDNA, although they had been shown to cleave the ssRNA equivalent to the same sequence.
Identification of the VapC target sequence
VapCPAE0151, VapCPAE2754, VapCRv0065, and VapCRv0617 all cleave 932 Pentaprobe RNA. Therefore, a set of short overlapping oligonucleotides covering 932 Pentaprobe RNA could be used as substrates for VapCPAE0151, VapCPAE2754, VapCRv0065, and VapCRv0617. Ribonuclease activity of VapC was tested against these short RNA oligonucleotides, and MALDI-TOF MS was used to identify the RNA cut sites. MALDI-TOF MS is a convenient tool for the analysis of many complex mixtures and is well suited to analysis of RNA molecules (during the ionization process DNA is prone to loss of bases, whereas RNA is more stable) (Nordhoff et al. 1993). Sample preparation is crucial for MALDI-TOF MS as chemicals commonly used in biochemical assays such as EDTA, glycerol, and MgCl2 can reduce sample ionization; RNA also forms adducts with sodium and potassium ions (Shaler et al. 1996), which decreases mass accuracy. It is therefore crucial for the sample to be desalted before MALDI-TOF MS analysis.
A variety of desalting techniques were trialed and assessed for their ability to minimize impurities, reduce adducts, and maximize sample recovery. All VapC proteins tested required NaCl and MgCl2 for activity. The use of cation exchange beads loaded with ammonium ions to exchange out the sodium ions was only moderately successful; sodium ion adducts were still present upon MS analysis (data not shown). C18 reverse-phase pipette tips, also known as ZipTips resulted in cleaner RNA, but the majority of the sample was lost in the process. Both lithium chloride and ethanol precipitation and sodium acetate and ethanol precipitation of RNA cleavage products were moderately successful approaches in precipitating small pieces of RNA, although some sample was still lost in the process and the use of sodium acetate did not remove sodium adducts from the RNA.
Ammonium acetate and ethanol precipitation of assay reactions resulted in maximum sample recovery, and the samples were more amenable to MALDI-TOF MS analysis. This method was successful for VapCPAE0151 and VapCPAE2754 assays as these are conducted in a Tris-HCl buffer. VapCRv0065 and VapCRv0617 assays were conducted in a sodium phosphate buffer giving rise to sodium adducts on the RNA. However, these VapC proteins demonstrated equivalent activity in an ammonium phosphate buffer. MALDI-TOF MS data were analyzed using in-house software that scanned all peaks and matched these with theoretical masses generated from the known oligonucleotide sequences.
This combination of Pentaprobes and MALDI-TOF MS of oligonucleotides based on a Pentaprobe RNA allowed us to readily characterize the sequence-specificity of VapC from the two organisms. VapCPAE0151 and VapCPAE2754 cleaved short RNA oligos based on the 932 Pentaprobe. Figure 4 shows the MALDI-TOF spectra of the cleavage products from a short 932-based oligo after incubation with VapCPAE0151 or VapCPAE2754. After 5 min, a large proportion of the RNA oligo is degraded, and after 60 min, all of the oligo is degraded and the higher-molecular-weight RNA fragments are also being degraded (Fig. 4A). This result suggests that there is an optimal recognition sequence for VapCPAE0151 and VapCPAE2754 and that other suboptimal sequences are also recognized for cleavage (Fig. 4B). Figure 4C summarizes the cleavage sites of VapCPAE0151 and VapCPAE2754 across all the short oligos tested. The MALDI-TOF MS results in Figure 4 confirmed that these two proteins target the same sequence and revealed the optimal cut site to be GG*UG, and also showed a less optimal cleavage site at GG*GG (Fig. 4C). Overall, it can be seen that a guanine residue is always positioned directly before the cleavage site, whereas there is some redundancy for the bases present at other positions in the target sequence (Fig. 4C).
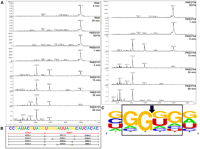
VapCPAE0151 and VapCPAE2754 target GGUG and GGGG sequences. (A) MALDI-TOF MS of 932 RNA oligonucleotide five negative controls (RNA 0 and 60 min and EDTA) show no degradation of the RNA oligonucleotide due to ribonuclease contamination. Time course assays of VapCPAE0151 (right panel) and VapCPAE2754 (left panel) show very little activity after 1 min at 37°C with VapC. After 5 min, a cut site at GGUG (red fragments in B) is observed with other less optimal cut sites appearing later in the time course (15–60 min; black fragments in B). (B) 932 RNA oligonucleotide five with corresponding cut sites and m/z values; dotted lines represent fragments that were below the MS detection limit. (C) Analysis of VapC cleavage sites across 932 RNA oligonucleotides as calculated by WebLogo (Crooks et al. 2004). The VapC cleavage position is indicated by an arrow. The height of the letter is proportional to the frequency of that base at that particular position.
VapCRv0065 and VapCRv0617 cleaved almost all of the short 932-based RNA oligos but with varying efficiency; analysis of the cleavage products for one of these short oligos by MALDI-TOF MS (Fig. 5A) revealed that VapCRv0065 and VapCRv0617 cut GC-rich 4mers, i.e., GGCG, GCCG, and GGGC (Fig. 5B). Analysis of the cleavage sites across all the RNA oligos cut by these two VapCs confirmed that VapCRv0065 and VapCRv0617 target GC-rich sequences (Fig. 5C). As with the other VapC proteins characterized in this study, there is redundancy in the target sequence and VapC will cleave a range of sequences less efficiently (Fig. 5C).

VapCRv0065 and VapCRv0617 target GC sequences. (A) MALDI-TOF MS of 932 RNA oligonucleotide three negative controls (RNA 0 and 60 min and EDTA) show no degradation of the RNA oligonucleotide due to ribonuclease contamination. Time course assays of VapCRv0065 and VapCRv0617 show optimal cut sites at GCC and GGG (indicated by red fragments in B). Other less optimal cut sites appear later in the time course (15–60 min; black fragments in B). (B) 932 RNA oligonucleotide three with corresponding cut sites and m/z values; dotted lines represent fragments that were below the MS detection limit. (C) Analysis of VapC cleavage sites across 932 RNA oligonucleotides as calculated by WebLogo (Crooks et al. 2004). VapC cleavage position indicated by arrow. The height of the letter is proportional to the frequency of that base at that particular position.
Table 1 lists the VapC proteins characterized to date and the range of sequence targets for these RNase enzymes. VapCRv0065 and VapCRv0617 cut GC-rich 4-mers, and VapCPAE0151 and VapCPAE2754 cut the G-rich sequences GGUG and GGGG. With all four proteins characterized to date, there is redundancy in the target sequences, in that they will cleave other sequences, albeit at a lower rate.
Sequence-specificity of VapCs characterized
Determination of the specific RNA sequence motif that is targeted by VapC proteins is just the first step in finding the biologically relevant target. It is likely that the biologically relevant target is a combination of sequence and RNA secondary structure. However, the identification of the target sequence is an important first step to characterizing the biological activity of individual VapC RNases.
Precise bonds cut by VapC
MALDI-TOF MS analysis also provides insight into the molecular mechanism of VapC RNase activity. The precise masses of the RNA cleavage products from VapCPAE2754, VapCPAE0151, VapCRv0065, and VapCRv0617 reactions reveal a 5′ phosphate present on the 3′ cleavage product. This contrasts with the reported mechanism of the MazF and PemK (Kid) toxins, as well as that recently described for VapCpMYSH6000 from S. flexneri and VapCLT2 from S. enterica. These enzymes have all been reported to cleave the phosphodiester bond to yield a free 5′ OH group on the 3′ cleavage product, and a 2′-3′ cyclic phosphate on the 3′ end of the 5′ cleavage product, similar to RNase H (Munoz-Gomez et al. 2005; Zhang et al. 2005; Winther and Gerdes 2011).
Our MALDI-TOF MS data are consistent with other metal-dependent ribonuclease enzymes that activate a water molecule to initiate nucleophilic attack and cleave the 3′-O-P bond of RNA to produce 3′ hydroxyl and 5′ phosphate cleavage products (Tadokoro and Kanaya 2009). Future work to establish the exact catalytic mechanism of VapC/PIN-domain proteins and the determinants of the sequence-specificity will allow prediction of the RNA sequences targeted by each member of this large family of proteins.
Kinetic analysis of VapC
Fluorogenic substrates have previously been used to analyze the reaction kinetics of ribonucleases (Park et al. 2001; Wang and Hergenrother 2007) and generally consist of single RNA bases flanked by short DNA sequences. These mixed oligonucleotides are labeled with a fluorophore at one end and a quencher at the other. When the fluorophore and quench are in close proximity, a small amount of fluorescence is observed. Upon cleavage of the substrate, the fluorophore and quench are no longer in close proximity, resulting in an increase in fluorescence. Based on this concept, we have developed a sensitive, continuous fluorometric assay to measure VapC activity.
VapCPAE2754 displayed no activity against a short, chimeric (ssDNA/RNA) oligonucleotide in which the cut site GGUG was flanked by six to seven DNA bases on each side (data not shown). This protein did, however, cleave the RNA equivalent of the chimeric oligonucleotide, suggesting that flanking RNA (but not DNA) sequences are required for catalysis. A substrate consisting of GGUG surrounded by randomized A, C, and U residues was designed to have only one cleavage site (Fig. 6A,B) for VapCPAE2754 (eliminating secondary cleavage). We added a fluorophore (6-FAM) to the 5′ end of this sequence and a quencher (IBkFQ) to the 3′ end, so that cleavage could be monitored by an increase in fluorescence at 518 nm. This strategy allowed determination of the Michaelis-Menten kinetics for VapCPAE2754 (Fig. 6C).
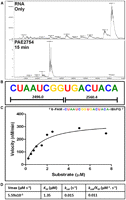
Design of a fluorometric substrate for determination of Michaelis-Menten kinetics of VapCPAE2754. (A) MALDI-TOF spectra of RNA oligonucleotide cleavage and (B) RNA oligonucleotide sequence showing only one cleavage site. (C) Reaction velocities were determined from the slopes of the fluorescent data sets and the calibration plot. The Michaelis-Menten equation was fit to the data for substrate concentrations 0.25–7.5 μM. (D) From the curve fit Vmax, KM and kcat were calculated.
CONCLUSIONS
We have developed a simple, effective, and general method for the isolation of toxic VapC proteins that can be applied to VapBC complexes from a range of organisms. Expression of soluble VapC from mycobacteria was only possible when in complex with their cognate antitoxins using M. smegmatis as an expression host. Then utilizing the inherent properties of VapB and VapC, the protease trypsin can be used to digest the less stable VapB, leaving active VapC. VapC can be subsequently purified from trypsin using anion exchange chromatography. This approach can be used routinely for VapBC complexes from M. tuberculosis.
The use of Pentaprobes and MALDI-TOF MS provides a new, robust method for determination of VapC sequence-specificity. Pentaprobes provide identification of cut sites of up to five bases in RNA molecules of shorter length, reducing the amount of secondary structure in the substrate. Shorter RNA oligonucleotides can then be designed to mimic regions of the whole Pentaprobe molecule and analyzed by MALDI-TOF MS to determine sequence-specificity. The sensitivity of this technique allows determination of the position of the phosphate moiety on the cleavage products and is useful for elucidation of RNA cleavage mechanisms. This method has been applied to four VapC proteins from two different organisms, identifying different cleavage sites for different proteins, outlining the effectiveness and versatility of the method.
The results presented here point to a number of possible biological roles for the vapBC operons in disparate bacteria. The G+C content of the two organisms considered in this work (51% for P. aerophilum and 66% for M. tuberculosis) suggests that the RNA target sequences for the VapC proteins (Table 1) are relatively common among the transcripts from each organism. This points toward VapCs acting as global down-regulators of mRNA transcripts under conditions where VapC is free of its cognate inhibitor VapB. It also seems likely that VapC will recognize not only sequence but also RNA secondary structure as a determinant of specificity, and this adds another layer of complexity to the determination of the cellular targets of VapC. Winther and Gerdes (2011) have recently shown that VapC from enteric bacteria specifically cleaves initiator-tRNA (presumably via a combination of sequence and secondary structure), affecting global translation when VapC is dissociated from VapB. The target sequences that we have identified rule out initiator-tRNA as a target for the VapC proteins from P. aerophilum and M. tuberculosis; however, it does not rule out VapC targeting tRNA molecules at other sites. Thus, our working hypothesis is that VapC targets a combination of RNA sequence and secondary structure, down-regulating specific RNA transcripts (or tRNA) in response to stress and thereby acting as post-transcriptional regulators of metabolism. Further in vivo experiments are required to determine the precise cellular targets for the VapCs from P. aerophilum and M. tuberculosis.
It is intriguing that VapCRv0065 and VapCRv0617 target the same RNA sequence. These two proteins are only distantly related, sharing just 22% sequence identity. These VapC proteins are part of an array of 46 VapC proteins in M. tuberculosis (Arcus et al. 2011). Intuitively, one might expect that distantly related VapCs would target unique subsets of RNA molecules. However, the evidence presented here suggests otherwise. This adds weight to the hypothesis that VapCs in M. tuberculosis are involved in global control of RNA in the cell under conditions where VapC is mobilized. This mobilization may be due to the up-regulation of cellular proteases under specific conditions that would readily degrade VapB and liberate VapC inside the cell.
MATERIALS AND METHODS
VapCPAE0151/VapCPAE0152 and VapCPAE2754 were expressed and purified according to the method described previously by Bunker et al. (2008) and Arcus et al. (2004), respectively. VapBC complexes Rv0065a-Rv0065 and Rv0617a-Rv0617 were cloned into pYUB28b according to the method described by Ahidjo et al. (2011) and expressed for the VapBC complex from M. smegmatis according to the method described by Robson et al. (2009).
Cloning of vapCMSMEG_1284 into pYUB1049
The open reading frame (ORF) encoding the vapC (MSMEG_1284) gene was amplified from M. smegmatis genomic DNA using primers engineered to contain an NcoI restriction site (underlined) in the forward primer (TAGCTGCCATGGTTATCGACACTTCTGC) and a BamHI restriction site (underlined) in the reverse primer (TATTTAGGATCCGCGTGGACCGCAGCG). The amplified products were digested, purified, and ligated into the pYUB1049 shuttle vector, enabling expression with a C-terminal His-tag. The construct was transformed into E. coli TOP10 cells and sequenced to ensure correct insertion. After sequencing, the constructs were transformed into M. smegmatis mc24517 electrocompetent cells and positive colonies selected by plating the transformants on 7H10 agar media supplemented with ADC (albumin, dextrose, catalase supplement), 0.05% (w/v) Tween 80, and 50 μg/mL kanamycin and hygromycin B.
Protein expression and purification using Mycobacterium smegmatis as a host
Small-scale expression tests were used to screen for expression of VapC in M. smegmatis. A single transformed colony was selected and used to inoculate a PA-0.5G/Tween 80 seeder culture and grown for 48 h at 37°C and diluted (1:100) into a ZYP-5052/Tween 80 expression culture. The expression culture was grown at 37°C, and aliquots of culture were taken at 24, 48, and 96 h and pelleted by centrifugation. Cell pellets were resuspended in 200 μL lysis buffer (50 mM phosphate buffer at pH 7.4, 200 mM NaCl, 20 mM imidazole). The cells were then lysed by sonication and the lysate centrifuged at 13,000g for 20 min to separate soluble and insoluble fractions. Samples were analyzed by SDS-PAGE for protein expression.
Plasmids were extracted by the alkaline lysis method and sequenced using T7 forward and reverse primers to investigate the absence of VapC expression.
Cloning of vapCMSMEG_1284 into pMAL-c2x
The ORF encoding the vapC gene (MSMEG_1284) was amplified from M. smegmatis genomic DNA using primers engineered to contain an EcoRI restriction site (underlined) in the forward primer (CTAGAATTCATGGTTATCGACACTTCTG) and a BamHI restriction site (underlined) in the reverse primer (TATGGATCCTCAGTGGACCGCAGC). The amplified products were then digested, purified, and ligated into pMAL–c2x, enabling expression of a MBP-VapC fusion protein. The construct was then transformed into E. coli BL21 (DE3) electrocompetent cells and plated on LB agar medium supplemented with 100 μg/mL ampicillin to select for positive transformants. Constructs were sequenced to ensure correct insertion.
Protein expression and purification of MBP-VapCMSMEG_1284 fusion protein
A single transformed colony was used to inoculate a rich media + glucose culture supplemented with 100 μg/mL ampicillin, which was then grown for 24 h at 37°C and was used at a 1:100 dilution to inoculate a rich media + glucose expression culture and grown at 37°C until an OD600 of ∼0.5 was reached. Protein expression was then induced with 0.3 mM IPTG, and cells were incubated for a further 2 h at 37°C for maximal protein expression.
E. coli expression cultures were pelleted by centrifugation prior to resuspension in 50 mM phosphate buffer (pH 7.4) 200 mM NaCl, and 1 mM EDTA and lysed by sonication. The cell lysate was centrifuged at 16,000g for 20 min to separate the soluble and insoluble fractions. The soluble fraction containing the MBP-VapC fusion protein was loaded onto a HiTrap MBP column (GE Healthcare). The column was washed with lysis buffer, and bound proteins were removed in a single elution step with 5 mL of elution buffer (50 mM phosphate buffer at pH 7.4, 200 mM NaCl, 1 mM EDTA, 10 mM maltose). Fractions containing the desired protein were analyzed by SDS-PAGE and subjected to either size exclusion chromatography or Factor Xa cleavage to remove the MBP protein from VapC. An initial evaluation of fusion protein cleavage was carried out using Factor Xa at 1%, 2%, and 3% (v/v) of the fusion protein. The reaction mixture was incubated for 2, 4, 6, or 24 h at room temperature (22°C) with shaking at 300 rpm. Low concentrations of SDS (0.01% and 0.05% [w/v]) were added to help improve cleavage of MBP.
Protocol for determining VapC ribonuclease activity and sequence-specificity
-
The Pentaprobe insert is PCR amplified using pcDNA3 forward (AGAGAACCCACTGCTTACTGGCT) and reverse (AGCGAGCTCTAGCATTTAGGTGACA) primers flanking the insert to include the T7 promoter. Pentaprobe RNAs are transcribed from the gel purified PCR product using the T7 MEGAscript kit (Ambion) and purified by sodium acetate and ethanol precipitation.
-
One microliter of purified VapC (1 mg/mL) is incubated with 1 μg of each Pentaprobe RNA for 60 min at 37°C in the presence of 6 mM MgCl2.6 Time points are taken and stopped by the addition of formamide loading dye and heated for 5 min to 70°C. Negative controls include the omission of VapC (RNA only), the addition of 12 mM EDTA, and the addition of 1 μL VapBC complex (1 mg/mL) instead of VapC. Samples are analyzed on 10% urea-denaturing polyacrylamide gels post-stained with SYBR Green II RNA stain (Invitrogen).
-
Overlapping RNA oligonucleotides are designed to model the Pentaprobe RNA that the VapC proteins of interest cleave and mimic the secondary structures of the equivalent regions on the whole Pentaprobe RNA.
-
Assay reactions containing 1 μL purified VapC (1 mg/mL) is incubated with 1 μg of each RNA oligonucleotide for 60 min at 37°C in the presence of 6 mM MgCl2 in a final volume of 10 μL. Assay reactions are stopped by addition of 5 M ammonium acetate (final concentration, 2 M) and three times the reaction volume of 100% ethanol and then chilled on ice for the remainder of the experiment (at least 30 min). The precipitated RNA fragments are centrifuged at 13,000g for 25 min at 4°C to pellet the RNA, and the pellet is washed with 70% ethanol and then resuspended in 10 μL nuclease free water (not DEPC treated).
-
Matrix for MALDI-TOF MS is prepared fresh and consists of 5 mg 3-hydroxypicolinic acid (3-HPA), 10 μL 2.5 M diammonium citrate, 125 μL acetonitrile (ACN), and 365 μL nuclease free water. The solution is vortexed well until all matrix is dissolved and then centrifuged at 13,000g for 5 min.
-
One microliter of matrix solution is spotted onto each spot on an Anchorchip target plate (Bruker Daltonics) and left to air dry. One microliter of oligonucleotide calibration standard (Bruker Daltonics) is then pipetted onto a matrix spot, and 1 μL of sample is pipetted onto subsequent spots and left to air dry.
-
Samples are analyzed on a MALDI-TOF mass spectrometer (Bruker Daltonics) in linear mode. Laser power is typically at ∼60%, but this can change depending on the ionization requirements of the sample. The mass range selector is set to low range, and a range of 2–12 kDa is collected (dependent on size of the oligonucleotide). The instrument is first calibrated with an oligonucleotide calibration standard (Bruker Daltonics) using an automatic polynomial correction before analysis of samples.
-
Spectra are saved and exported to the DataAnalysis software (Bruker Daltonics), peaks identified and labeled, and the baseline subtracted.
-
The mass list and corresponding intensities are exported into a comma separated excel file. The data are then processed using in-house software to determine possible cut sites. The intensity cut off was set to more than 1400 so all peaks above 1400 intensity will be picked for analysis. The software determines the difference between the predicted mass of the oligonucleotide and the actual value as measured by MALDI-TOF MS. The fragments with the closest matching masses will determine the cut site.
Kinetic analysis of VapCPAE2754
Fluorogenic substrate design
A chimeric RNA/DNA oligonucleotide was designed with four RNA bases flanked by DNA (5′-TAAGTCrGrGrUrGACATCAG-3′) and tested for ribonuclease activity by MALDI-TOF MS as per the protocol above. The RNA oligonucleotides UAAGUCGGUGACAUCAG and CUAAUCGGUGACUACA were tested for single cut sites by MALDI-TOF MS as above. The CUAAUCGGUGACUACA oligonucleotide labeled with 6-FAM (6-carboxyfluorscein fluorophore) at the 5′ end and IABkFQ (iowa black flurophore quencher) at the 3′ end was ordered from Integrated DNA Technologies (IDT) along with the cleavage products, 6-FAM-CUAAUCGG and UGACUACA-IABkFQ.
Calibration plot
Reactions containing a 1:1 molar ratio of the labeled 5′ and 3′ cleavage products at various concentrations (0.1, 0.25, 0.5, 1, 1.25, 1.5 μM), 12 mM Tris-HCl, 12 mM NaCl, 6 mM MgCl2, to 300 μL with nuclease free water were equilibrated to 37°C. Fluorescence was measured as described above for 100 sec. Fluorescent values were used to construct a calibration plot of relative fluorescent units (RFU) versus oligonucleotide fragment concentration.
Fluorogenic substrate cleavage assay
A Hitachi F-700 fluorescence spectrometer (Hitachi High Technologies) was fitted with a Hitachi Thermostatted cell holder accessory. The cell was maintained at 37°C by water feed from a water bath at 5.8 L/min. Time scans used a 0.5 data pitch, with PMT set to 450 V, excitation (ex.) 485 nm, emission (em.) 518 nm, 5 nm slits, and 2 msec response time. Scans for assays and blanks were recorded for 100 sec.
A stock VapCPAE2754 was prepared at 0.2 mg/mL and a stock substrate solution prepared to 100 μM stock in nuclease free water. Three hundred microliter assay reactions containing various concentrations of fluorogenic substrate (0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 5.0, and 7.5 μM), 12 mM Tris-HCl, 12 mM NaCl, 6 mM MgCl2 were pre-equilibrated to 37°C and the blank read for 100 sec before addition of 10 nM VapCPAE2754 and fluorescence subsequently measured for 100 sec.
Kinetic analysis
Slopes derived from VapCPAE2754 cleavage of the fluorogenic substrate were compared with controls where no enzyme was added. The control reactions exhibited no change in fluorescence over the assay time course and, therefore, were not subtracted from the data obtained after addition of VapCPAE2754. The RFU for the assay reaction was converted to micromoles oligonucleotide cleaved using the slope from the calibration plot. Graphpad Prism was used for nonlinear regression analysis on these data to obtain reaction velocities for a range of substrate concentrations. The Michaelis-Menten equation was then fit to the data.
ACKNOWLEDGMENTS
This research was funded by the Health Research Council New Zealand and the Marsden Fund, Royal Society of New Zealand. J.L.M. was funded by a PhD scholarship from the University of Waikato.
Footnotes
-
↵5 Corresponding author.
E-mail varcus{at}waikato.ac.nz.
-
↵6 Ribonuclease activity assays for VapCRv0065 and VapCRv0617 from M. tuberculosis were conducted in 12 mM phosphate buffer (sodium phosphate typically used except for MALDI-TOF MS where ammonium phosphate was used), 6 mM NaCl, and 6 mM MgCl2. Ribonuclease activity assays for VapCPAE0151 and VapCPAE2754 from P. aerophilum were conducted in 12 mM Tris.HCl, 6 mM NaCl, and 6 mM MgCl2. Individual assay reactions were set up for each time point to reduce the possibility of RNase contamination.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031229.111.
- Received November 6, 2011.
- Accepted February 26, 2012.
- Copyright © 2012 RNA Society








